本文是一篇临床医学论文,笔者经过研究,得出结论:PPARγ在IgG-IC肺内沉积后的抗炎作用是通过抑制Egr-1的产生来介导的,Egr-1可以与包括TNF-α、MCP-1和MIP-1α在内的炎症因子的启动子区域结合。PPARγ激动剂罗格列酮对PPARγ-Egr-1-炎症因子轴的调节可能代表了一种治疗急性肺损伤的新策略。
第一章综述
1.1 IgG-IC诱导急性肺损伤
1.1.1 IgG-IC诱导急性肺损伤概况
ALI/ARDS是指急性呼吸衰竭的临床症状,ARDS是一种急性弥漫性炎症性肺损伤,导致肺血管通透性增加、肺的湿/干重比增加和肺泡结构被破坏[6],发病率高[7]。临床特征是低氧血症和双侧影像学肺纹理增粗出现斑片影甚至有大片状病灶,生理死腔增加,这些都与肺顺应性降低有关[8]。当ARDS处于急性期时,形态学特征为弥漫性肺泡损伤,包括水肿、炎症反应、透明膜病变甚至出血这几种现象。ARDS是一个重要的全球公共卫生问题,具有地域差异和大约40%的极高死亡率[1]。ALI可由严重败血症、严重细菌性肺炎、外伤、烧伤等事件诱发[9]。已有大量证据证明,ALI/ARDS与IgG-IC沉积有关[2]。由IgG-IC沉积引发的肺部炎症反应虽然不是肺部独有的,但可用于量化和评估肺部炎症水平。在动物实验中,急性肺损伤可由肺血管壁中免疫复合物的沉积或由注入气道的免疫复合物所触发,IgG-IC诱导的小鼠急性肺损伤模型是急性肺损伤的经典模型,啮齿动物肺中的IgG-IC沉积模型已被广泛用于研究肺组织炎性损伤。
1.1.2 IgG-IC诱导急性肺损伤的发病机制
IgG-IC诱导ALI/ARDS的过程中,局部巨噬细胞首先通过Fcγ受体与配体的结合被激活,产生并分泌补体激活产物C5a和大量炎症因子包括肿瘤坏死因子α(TNF-α)和白细胞介素-1(IL-1)以及趋化因子。然后,这些细胞因子引起肺血管内皮上粘附分子的上调,包括细胞间粘附分子-1(ICAM-1)以及P-和E-选择素(P-and E-selectins),从而增强中性粒细胞的粘附性,促进它们向肺间质和远端气道转移。肺泡上皮细胞ICAM-1的表达增加了肺泡巨噬细胞对上皮细胞的粘附性,从而增加了细胞因子和趋化因子的产生,包括CXC趋化因子和CC趋化因子。CXC趋化因子,如巨噬细胞炎症蛋白-2(MIP-2)和细胞因子诱导的中性粒细胞趋化剂(CINC)在肺中产生形成一个化学引诱梯度,招募中性粒细胞进入肺。CC趋化因子,如MIP-1α和MIP-1β,可作为肺泡巨噬细胞的自分泌激活因子,促进急性炎症进展[10,11]。
..............................
1.2过氧化物酶体增殖物激活受体γ(PPARγ)
1.2.1 PPARγ简介
PPARs是配体调节转录因子的核受体超家族的成员[3,18]。在哺乳动物中,有三种PPARs:PPARα(也称为NR1C1),PPARβ/δ(也称为NR1C2)和PPARγ(也称为NR1C3)[19-22]。PPARs的配体包括多种脂肪酸或脂肪酸衍生物,通过配体受体结合活化实现对下游的各种分子调控作用[19-22]。PPARs的结构与典型的核受体结构相似,由不同的功能域组成,包括一个N末端反转录激活域(AF1)、一个高度保守的DNA结合域(DBD)和一个C端配体结合域(LBD),其中包含一个配体依赖的反转录激活功能(AF2),多个结构域可协同调节受体的功能。它们通过DBD中的两个锌指与DNA序列特异性结合,并通过LBD中的配体结合口袋来结合配体。DNA的结合需要与类视黄醇X受体(RXR)家族成员发生二聚化,然而,PPAR和RXR之间的异二聚化与配体无关,依赖于这两种受体的LBD和DBD中的异二聚化界面[24-26]。
PPARγ在人体内各种组织中广泛表达,而在白色脂肪组织(WAT)和棕色脂肪组织(BAT)中表达最多,它是脂肪形成的主调节因子,也是全身脂质代谢和胰岛素敏感性的有效调节因子。PPARγ/RXR异二聚体是当配体结合活化PPARγ后再与RXR结合形成的,该异二聚体通过与靶基因启动子上游的PPAR反应元件(PPRE)结合,进而调控靶基因的表达,这些靶基因参与了体内的脂质代谢、免疫反应、和细胞稳态的维持[24-26]。PPARγ不仅可以作为转录激活剂,还可以抑制靶基因的表达。有研究表明,PPARγ的过表达与PPARγ激动剂联合使用会导致成纤维细胞中某些基因的表达显著降低[24-26]。有趣的是,成熟脂肪细胞中的某些脂肪细胞基因在脂肪生成过程中被PPARγ正向调控,但是合成的PPARγ激动剂却下调了它们的表达[28-30]。
..............................
第二章实验器材与方法
2.1实验仪器与耗材
2.1.1实验仪器

临床医学论文参考
.............................
2.2实验方法
2.2.2细胞复苏
(1)从-80℃冰箱中取出用无血清细胞冻存液冻存的细胞,提前打开37℃水浴槽,将冻存管进行解冻。
(2)待细胞混合液完全融化后,在超净台中立即将1ml细胞培养基加入管内,用移液枪混匀,将混合液转移到5ml的培养基中,1000rmp,5min离心后用真空泵吸去上清,保留底层的细胞沉淀,操作过程应轻柔,避免将细胞沉淀吸走。
(3)用移液枪取培养基将细胞沉淀轻柔吹打混匀为细胞悬液,取20µl加入细胞计数板中,使用细胞计数器计数,计数后调整适宜的细胞密度,将细胞混合液移至事先准备好的培养皿中,放入细胞培养箱中培养。
(4)于24h后换液,继续培养至形成单层贴壁细胞后传代。
2.2.3细胞培养和传代
(1)提前在超净台内配置好RAW264.7细胞和HEK293细胞需要的的培养基,4℃冰箱保存。
(2)在显微镜下观察6cm培养皿中RAW264.7细胞和HEK293细胞生长情况,待细胞达到一定密度后进行消化传代。
(3)用台式真空泵吸去变黄的培养基,加入1ml1×PBS,轻柔摇晃数次后清洗贴壁细胞表面残留液体,用真空泵吸去PBS。
(4)加入适量胰酶足以覆盖贴壁细胞,胰酶不宜过多过少,放入培养箱中消化,RAW264.7细胞消化时长大约30min,HEK293细胞消化时长为3-5 min,用1ml细胞培养基将贴壁细胞吹落成细胞悬液,收集起来。
(5)4℃3500rpm离心1min后,去除上层的胰酶消化液,注意控制真空泵的吸力,切勿吸到细胞(每次使用真空泵时替换新的枪头吸引,避免污染)。
(6)用1ml培养基吹打底部细胞沉淀制成单细胞悬液,取300µl至提前加入3mL培养基的培养皿,细胞培养箱中培养(定期给细胞换液,换液频率视细胞密度和状态而定)。
...............................
第三章实验结果................................25
3.1实验结果一:PPARγ负调控IgG-IC诱导的急性肺损伤.....................25
3.1.1 PPARγ在IgG-IC诱导的急性肺损伤中表达量变化................................25
3.1.2 PPARγ在IgG-IC诱导的急性肺损伤中的作用................................25
第四章讨论...............................38
第三章实验结果
3.1实验结果一:PPARγ负调控IgG-IC诱导的急性肺损伤
3.1.1 PPARγ在IgG-IC诱导的急性肺损伤中表达量变化
在IgG-IC诱导的急性肺损伤模型中,PPARγ表达水平和具体功能尚未确定。因此,首先需要研究了IgG-IC在肺内沉积后,小鼠肺组织中PPARγ的表达水平是否发生了改变。如结果所示,qPCR检测了IgG-IC处理后导致小鼠肺组织中PPARγ的mRNA水平下降36%(图1A),该结果与Westernblot显示的蛋白水平变化结果一致(图1B)。
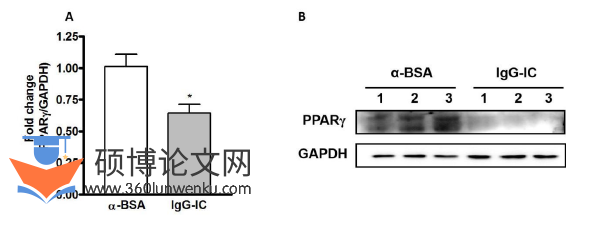
临床医学论文怎么写
3.1.2 PPARγ在IgG-IC诱导的急性肺损伤中的作用
PPARγ在IgG-IC诱导的急性肺损伤中的作用可通过检测肺通透性和MPO活性(MPO是中性粒细胞的功能标志和激活标志)来评估急性肺损伤程度。首先,利用腺病毒作为载体,在小鼠肺组织中过表达PPARγ,发现即使在1×108 PFU(斑块形成单位)的剂量下,Ad-PPARγ处理也能显著增加PPARγ的表达量(图1C)。因此,在接下来的研究中,我们将1×108 PFU的Ad-PPARγ应用于动物实验模型中。与对照组相比,IgG-IC刺激后,肺通透性指数增加了2.5倍以上,而PPARγ过表达的情况下,肺通透性指数几乎降低到基础水平(图1D)。与预期的一样,IgG-IC处理引起的肺MPO活性的增加同样也被PPARγ过表达显著抑制(图1E)。也就是说在IgG-IC诱导的急性肺损伤中,PPARγ可以减少肺通透性并降低MPO活性,有一定的缓解肺损伤的作用。
...................................
第四章讨论
ARDS是呼吸衰竭的主要原因,它约占ICU入院人数的10%[111]。分析严重急性呼吸衰竭全球影响的大型观察性研究(LUNG-SAFE研究)表明,这种综合征既未得到充分认识,也未得到充分治疗,并且与高死亡率相关[113]。由于临床上缺乏特效药,美国ALI/ARDS的死亡率曾经甚至高达60%[114,115],即使采用呼气末正压(PEEP)和肺复张策略(LRM)等肺部保护性策略[116],死亡率仍然高达40%[113],目前疾病进展形势仍然令人担忧,是一项重大的临床挑战。因此,我们需要进一步研究ALI/ARDS的分子机制,以帮助寻找新型有效的治疗方式。ARDS/ALI的病因多样且发病机制复杂,IgG-IC肺内沉积也是其中一种重要的病因,在ARDS/ALI患者的肺泡灌洗液中也可检测到IgG-IC,由IgG-IC的肺内沉积构建的啮齿动物肺损伤模型可诱导广泛的基因激活,并已用于研究细胞因子、趋化因子和补体在急性炎症过程中的作用[113],因此我们利用IgG-IC诱导的急性肺损伤模型来揭示急性肺损伤的潜在机制。既往的各种研究已经证明,IgG-IC刺激可引起明显的急性炎症反应,其特征是中性粒细胞浸润到肺组织中,细胞因子和趋化因子的表达升高,这与临床症状相似。我们通过实验发现PPARγ-Egr-1-炎症因子轴参与了IgG-IC诱导的肺损伤,而对该轴的干预可能是治疗ALI/ARDS患者的有效策略,为完善治疗策略提供新思路。
肺内IgG-IC沉积后,局部巨噬细胞表达高水平的Fcγ受体,然后巨噬细胞释放TNF-α和IL-1β等细胞因子,这对刺激肺内皮细胞表达粘附分子至关重要[118,119]。此外,参与早期反应的细胞因子例如TNF-α,IL-1-β和补体激活产物C5a促进肺组织中趋化因子包括CXC趋化因子,例如MIP-2和CINC,还有CC趋化因子例如MIP-1α和MCP-1[120-122]。综上所述,肺损伤和大量中性粒细胞进入肺组织是由多种炎症相关介质的联合作用引起的,包括细胞因子、趋化因子和粘附分子。实际上,上述所有过程都是由各种转录因子组成的网络控制的。此前研究表明,在IgG-IC诱导的急性肺损伤过程中,NF-κB和AP-1的DNA结合活性增加[120-122]。这两种转录因子都可以直接结合到炎症因子的启动子区域,并增加它们的表达量。此外,我们实验室的研究结果表明,C/EBPb和C/EBPd,,都属于C/EBP家族成员,都可以通过结合炎症基因启动子区域中的C/EBP组件来促进IgG-IC引发炎症反应[120-122]。在本研究中,我们发现在IgG-IC处理后,PPARγ的表达量显著下降。然而,作为一个重要的转录因子,PPARγ在IgG-IC引发的炎症中的作用尚未确定,它与炎症因子之间的桥梁分子有待探究,其中复杂的网络关系需要完善。
参考文献(略)