前 言
牙周炎是牙周支持组织的慢性感染性疾病,临床上主要表现为附着丧失、牙周袋形成及牙槽骨吸收。慢性牙周炎的始动因子是菌斑微生物的形成,但软硬组织的破坏则是由微生物和宿主的免疫应答共同介导的。而目前临床上所应用的治疗手段主要是针对菌斑微生物的去除,针对宿主免疫应答的治疗仍处于探索阶段。HPDLF 作为牙周膜中分布最多,功能最重要的细胞,一直是牙周炎发病机制研究中的重点。越来越多的证据表明,HPDLF 具有部分类似固有免疫细胞的免疫功能:表达模式识别受体、分泌细胞因子及趋化因子、活化 T、B 淋巴细胞等。通过课题组前期研究发现,在牙周炎组织中 UPR 相关分子表达升高,同时炎症来源的牙周膜干细胞 UPR 通路处于活化状态。鉴于 UPR 在多种疾病中与炎症的发生发展存在着密切的联系,了解 HPDLF 的 UPR 状态,以及 UPR 对于细胞免疫功能的影响,可以为以 HPDLF 为靶点的免疫治疗提供新的研究依据。鉴于此,本研究中,我们以 HPDLF 对于巨噬细胞 M1/M2 极化的调节功能为切入点,探讨 UPR 对于 HPDLF 固有免疫相关功能的影响及其可能的机制;另一方面,我们以巨噬细胞表面抑制性分子LAIR-1对于巨噬细胞M1/M2极化的影响为切入点,试图证实 HPDLF 的 UPR 状态与单核巨噬细胞表面 LAIR-1 的表达之间是否存在联系。为此,我们设计以下三部分实验:1、 通过比较 P-HPDLF 与 H-HPDLF 中 UPR 相关分子的表达,了解炎症微环境中 HPDLF UPR 是否处于活化状态;体外使用 LPS、炎性细胞因子 TNF-α、IL-1β分别刺激 H-HPDLF,检测其 UPR 水平,进一步明确 HPDLF 炎症状态与 UPR 的相关性。2、 通过将 LPS 预处理的 HPDLF 与单核巨噬细胞共培养,检测巨噬细胞M1/M2 极化水平,以明确 TLR-4 活化后的 HPDLF 是否调节以及如何调节巨噬细胞的极化状态;使用 LPS 作用已经处于 UPR 激活状态的 HPDLF,检测 IL-1β、TNF-α、IL-6、IL-8 的分泌水平、TLR4 表达水平、NF-κB 通路的活化水平、及其对于对巨噬细胞极化的影响,使我们从模式受体的表达、细胞因子的分泌、炎症相关通路的活化以及对巨噬细胞的调控四个方面了解 UPR 对 HPDLF 的免疫功能的影响。3、 我们推测 UPR 状态的 HPDLF 可能通过调节巨噬细胞表面抑制性受体LAIR-1 分子的表达进而影响巨噬细胞的极化。并且从以下两个个方面试图证实我们的推测:HPDLF 的 UPR 状态是否影响巨噬细胞 LAIR-1 的表达;LAIR-1 的活化是否影响巨噬细胞极化。
..........
文献回顾
1.牙周膜成纤维细胞的免疫调节功能
慢性牙周炎是菌斑微生物作用下牙周支持组织发生的慢性炎症性疾病。菌斑生物膜的形成是牙周炎症的始动因子,其毒力成分可以直接破坏牙周组织;但宿主对微生物的免疫应答,包括固有免疫应答(innate immune response)和适应性免疫应答(adaptive immune response)才是牙周支持组织破坏的主要原因。外源性病原微生物入侵牙周组织时,固有免疫应答会第一时间启动,抵抗病原微生物入侵,并通过表达共刺激分子、分泌细胞因子调节适应性免疫应答。固有免疫细胞主要包括单核-巨噬细胞、树突状细胞、中性粒细胞、自然杀伤细胞、肥大细胞等,可以通过其胞膜及胞浆内表达的特异性受体识别相应配体,快速启动固有免疫应答。这些受体统称为模式识别受体,可以识别不同类型微生物共同表达的高度保守结构,即病原体相关分子模式,这些保守结构表达于病原体及被病原体感染的细胞表面,是固有免疫应答的强激活剂。此外,模式识别受体还可以通过识别凋亡细胞表面呈现的凋亡细胞相关分子模式(apoptotic cell associated molecular pattern,ACAMP)清除凋亡细胞。常见的模式识别受体包括表达在细胞表面及胞内的 Toll 样受体;表达在细胞膜上的C 型凝集素受体(CLR);表达在胞质的 NOD 样受体(NLRs)和 RIG-/I 样受体(RLR)等。随着对于固有免疫认识的深入,逐步发现一些非免疫细胞除了参与固有免疫屏障的构成外也具有一些类似固有免疫细胞的功能,包括表达模式识别受体、识别外源性抗原、分泌多种细胞因子和趋化因子等,同时对固有免疫和适应性免疫细胞具有调节作用。牙周膜成纤维细胞是牙周膜中数量最多、功能最重要的细胞,其最主要功能是参与细胞外基质的合成和代谢[3, 4]。近年来的研究表明牙周膜成纤维细胞同样具有上述类似固有免疫细胞的功能[5]。
.........
1.1 牙周膜成纤维细胞表达多种模式识别受体
大量研究已经证实牙周膜成纤维细胞表面表达 TLR2 和 TLR4[6]。TLR4 可以识别革兰氏阴性菌细胞壁成分脂多糖,TLR4 通过其胞膜外区识别 LPS,通过胞内的Toll-IL-1 受体结构域(TIR)将信号传递至细胞内,从而引发一系列细胞免疫反应。TLR2 主要识别革兰氏阳性菌胞壁上的脂蛋白和肽聚糖。在牙龈卟啉单胞菌(Porphyromonas gingivalis, P.g)作用下,牙周膜成纤维细胞表面的 TLR2/4 数量增多并且发生活化[7],活化信号传递到胞内可以促进多种细胞因子及趋化因子的分泌[8,9];促进基质金属蛋白酶(MMP)的产生[10]。NLR 家族成员核苷酸结合寡聚化结构域 1 (Nucleotide-binding oligomerization domain, NOD1)、NOD2 同样被证实表达于牙周膜成纤维细胞[11]。iE-DAP(NOD1 ligand)或 MDP(NOD2 ligand)作用细胞后,可以增加牙周膜成纤维细胞炎性细胞因子及趋化因子的分泌,进而促进白细胞向牙周组织迁移及黏附[12]。Liu 等指出 P.g 可以增加牙周膜成纤维细胞胞浆内的 NOD1/2的表达[13];并证实 P.g 诱导细胞产生 IL-6、IL-8、VCAM-1、ICAM-1 的过程依赖NOD1/2 的参与[13, 14]。而下调牙周膜成纤维细胞 NOD1/2 和 TLR4 表达后,其与单核-巨噬细胞的黏附受到明显抑制[15]。在正常牙周膜成纤维细胞及炎症的牙周组织中均有 NLRP3 的表达,胞壁酰二肽(Muramyl Dipeptide, MDP)及 LPS 均可使牙周膜成纤维细胞内 NLRP3 的表达增加[16];同时也已经证实 NLRP1 和 NLRP3 炎症小体的活化参与了牙周膜成纤维细胞 IL-1β 的分泌[16, 17]。
.........
1.2 牙周膜成纤维细胞通过分泌细胞因子及趋化因子参与固有免疫和获得性免疫的调节
肿瘤坏死因子 α(Tumor necrosis factor, TNF-α)一种强效单核因子。其他如 T淋巴细胞、B 淋巴细胞、中性粒细胞及内皮细胞等在一定条件下亦可产生。TNF-α作用于内皮细胞,可增加其表面黏附分子如 ICAM-1、VACM-1、ELAM-1 等的表达,同时损伤内皮细胞并增加血管通透性;另一方面,TNF-α 可以诱导单核细胞、淋巴细胞表面黏附分子的表达和构象发生变化,最终导致单核细胞和中性粒细胞的黏附和游出。TNF-α 还可以促进 T 细胞的杀伤功能,提高中性粒细胞的吞噬功能。牙周膜成纤维细胞在细菌脂多糖、正畸力等作用下可以分泌 TNF-α[18, 19]。炎症来源的牙周膜成纤维细胞中 TNF-α 水平明显增高[20]。TNF-α 可以增加巨噬细胞中RANKL/OPG 的比率,促进破骨细胞分化,同时增强破骨细胞活性[21];可以诱导牙周膜成纤维细胞产生基质金属蛋白酶,破坏胶原纤维,降解细胞外基质[22, 23];同时抑制细胞碱性磷酸酶的活性,抑制成骨分化[24]。TNF-α 还可以通过活化单核巨噬细胞,促使其分泌 IL-1、IL-6、IL-8 等细胞因子。而在动物牙周炎模型中,局部注射TNF-α 拮抗剂可以明显减少骨吸收水平[25]。但也有证据显示,在牙周膜成纤维细胞和外周血单个核细胞(PBMC)共同培养体系中,虽然 TNF-α 浓度增加,但 TNF-α并没有参与牙周膜成纤维细胞诱导巨噬细胞破骨分化的过程[26]。白细胞介素 1β(interleukin-1β,IL-lβ)同样是一种强效的单核因子,其它如树突状细胞、成纤维细胞、内皮细胞和上皮细胞等在一些特定条件下也可以产生IL-1β[27]。牙周膜成纤维细胞在细菌脂多糖、尼古丁、机械力等作用下均可产生IL-1β[28,29];与 PBMC 直接或间接共培养亦可促进其分泌 IL-1β。IL-1β 作用于单核巨噬细胞、树突状细胞、T 细胞、中性粒细胞、嗜酸性粒细胞,可以促使这些细胞活化并释放大量的细胞因子和趋化因子;作用于牙周组织中的牙周膜成纤维细胞及牙龈成纤维细胞,促进其前列腺素 E2、基质金属蛋白酶、胶原酶的分泌,引起胶原的降解、软组织附着丧失及骨吸收;可以增加内皮细胞表面粘附分子的表达,促进淋巴细胞黏附并穿出血管内皮,在炎症局部聚集浸润;也有报道已经证实 IL-1β 可以通过促进巨噬细胞的破骨分化,参与牙骨质和牙槽骨的吸收过程。在牙周炎动物模型中,治疗性的阻断 IL-1 受体可以明显减少局部炎症细胞的浸润、破骨细胞的活化以及骨吸收的水平[30, 31]。
..........
2 巨噬细胞极化参与牙周炎症的发生发展
巨噬细胞可以根据其活化类型和功能分为经典激活的 M1 型和旁路激活的 M2型。M1 型巨噬细胞可由 γ 干扰素(IFN-γ)单独激活也可由 IFN-γ+LPS 或者IFN-γ+TNF-α 共同激活。M1 型巨噬细胞分泌大量促炎细胞因子,如 IL-1β、TNF-α、IL-12、IL-23、IL-27,不分泌或极少分泌 IL-10;表达 iNOS 和精氨酸;其细胞表面表达 MHCⅡ、TLR 以及共刺激分子 CD86;分泌趋化因子 CXCL9、CXCL10;参与Th1 及 Th17 细胞炎症反应。通过旁路激活途径 IL-4、IL-13、IL-10 和免疫复合物等可诱导形成 M2 型巨噬细胞,主要分泌 IL-10、IL-4、IL-1RA,几乎不分泌 IL-12、IL-23;表达高水平的精氨酸酶;其细胞表面表达高水平的清道夫受体、半乳糖受体和甘露糖受体[45],分泌趋化因子 CCL17、CCL22、CCL24 与 Th2 细胞表面 CCR3 CCR4 结合[46-48],促进组织修复;同时分泌转化生长因子 β、表皮生长因子、血管内皮生长因子等促进局部血管及细胞外基质的改建和生成。巨噬细胞 M1/M2 极化是巨噬细胞分化过程的两个方向,而非将巨噬细胞进行绝对的分型,而且在一定组织微环境作用下可以相互转化,同时在 M1、M2 之间存在着多种中间型,其中最主要的是以分泌IL-10 为主的调节性巨噬细胞,其他中间型例如肿瘤相关巨噬细胞同时表达 M2 和调节性巨噬细胞特质;肥胖相关巨噬细胞则同时表达 M1 和 M2 特质。这些非经典激活的中间型都被归为 M2 分型。
........
第二部分 UPR 状态对 HPDLF 免疫相关功能的影响....... 44
1. 材料........ 44
2. 方法........ 46
3. 结果........ 50
4. 讨论........ 65
第三部分 UPR 状态 HPDLF 对巨噬细胞表面抑制性分子 LAIR-1 表达的影响....68
1. 材料........ 68
2. 方法........ 69
3. 结果........ 70
4. 讨论........ 76
第三部分 UPR 状态 HPDLF 对巨噬细胞表面抑制性分子 LAIR-1 表达的影响
LAIR-1 是表达于大部分免疫细胞表面的一种免疫抑制性受体。体外研究表明,LAIR-1 在经过单克隆抗体交联活化后,向胞内传递强烈的抑制性信号,通过去磷酸化抑制包括自然杀伤细胞、T 淋巴细胞、B 淋巴细胞和树突状细胞等多种免疫细胞的功能。但关于 LAIR-1 与单核巨噬细胞极化的相关研究国内外尚无报道。LAIR-1 被证实与多种自身免疫性疾病相关,其中研究最多的是类风湿性关节炎和系统性红斑狼疮。LAIR-1 被证实高表达于 RA 患者关节腔内的巨噬细胞及滑膜成纤维细胞,并且可以抑制破骨细胞的分化。牙周炎作为一种由微生物和宿主反应共同介导的,以骨吸收为主要症状的慢性炎症,与 RA 有着许多相似的发病机制,胶原的异常代谢、单核巨噬细胞的极化和破骨细胞的分化都在这两种疾病的发展中起到重要的作用。但目前尚无有关 LAIR-1 与牙周疾病的相关报道。
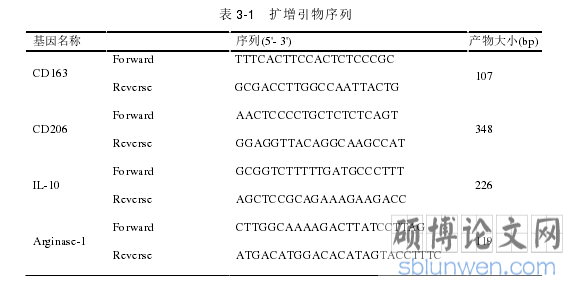
........
小结
第一部分:本实验首次通过比较 H-HPDLF 与 P-HPDLF,证明 P-HPDLF 中 UPR处于活化状态,通过体外模拟炎症微环境,进一步证实 LPS、IL-1β、TNF-α 均可诱导 HPDLF 发生 UPR。
第二部分:通过我们的实验比较完整的展现了牙周膜成纤维细胞的免疫功能,包括表达模式识别受体、分泌细胞因子及趋化因子、调节巨噬细胞功能。同时我们证实了 UPR 状态的细胞,其对于 LPS 的反应性出现明显的下降。表现为 UPR 状态的HPDLF 在 LPS 作用后,细胞表面 TLR4 的增加受到抑制,细胞内炎症相关通路 NF-κB的活化受到抑制,细胞培养上清中 IL-1β、TNF-α、IL-6 和 IL-8 的浓度显著降低,同时 HPDLF 对于单核巨噬细胞 M1 极化的促进作用受到抑制。
第三部分:牙周致病因子 LPS 及牙周炎组织中高表达的细胞因子 TNF-α、IL-1β分别作用单核巨噬细胞,细胞表面抑制性受体 LAIR-1 表达水平明显降低,这是在牙周炎领域首次关于该分子的报道;经 LPS 预处理的 HPDLF 同样具有降低巨噬细胞LAIR-1 的作用,而 UPR 状态 HPDLF 对 LAIR-1 的降低作用较弱,说明 HPDLF 的UPR 状态对于 LAIR-1 的降低有一定的保护作用。同时我们也首次证明巨噬细胞表面LAIR-1 参与抑制巨噬细胞 M1 极化。
..........
参考文献(略)