本文是一篇计算机论文,本文提出了一种用于分子属性预测的注意力编码方法。本文的注意力编码方法使用已知分子表示未知分子,并将未知分子原来的表示和已知分子对其进行的表示拼接成一个向量,输入到一个前馈神经网络中进行映射。本文认为,这种编码方法较为简单,也许还存在更好的编码方法,但使用已知样本加深对未知样本的认识这一想法必然能产生更大的反映。
第一章 绪论
1.1 研究背景与研究意义
分子属性预测,即给出一个分子,预测其是否具备某种属性。分子属性预测在定量化学、药物发现、材料发现等领域中具有非常重要的意义[1-4]。例如,在药物发现中,通常遵循这样的流程,首先科学家们从一堆分子中挑选出一些分子,使用这些分子进行药物的开发;在药物开发完成后,在动物的身上进行实验,以观测毒性、药代等新药的多种性质;之后通过申请后进行临床研究,一般需要三四期的临床研究;在一切都没有问题后新药才能投放到市场。因为无法确定新药具备的性质(是否具有毒性等),所以很多新药研发工作走不到临床。一些研究显示,一个药物发现需要花费近10年的时间,20亿美元,且临床成功的概率只有10%左右[5-7]。一旦我们能够知道目标分子对应的属性,便能够加快药物、材料发现的过程,并节省大量资源。
密度泛函理论(DFT)[8-9]在分子的物理属性预测中扮演着重要的角色。密度泛函能够计算分子的能隙、电子的激发态、分子和原子之间的范德华力等,但密度泛函这一方法存在一些缺陷,首先密度泛函理论不能计算分子的化学性质,其次,密度泛函这一方法需要极高的计算时间花费[10-11],例如,计算一个仅含有17个原子的分子的属性就需要超过1小时的时间[11]。显然,在药物发现这样需要从大规模分子空间中挑选出合适的一些分子,使密度泛函的应用受到了极大的限制。所以,寻找比密度泛函更有效的方法成为科研人员的目标。
近年来,被机器学习特别是深度学习在计算机视觉、自然语言处理等领域显著的成果所鼓舞,科研人员使用机器学习方法预测分子的属性[1,11-17]。然而,机器学习特别是深度学习,需要建立在大规模数据的基础上才能获得有效的模型,例如被广泛用于图像识别、图像分类的Imagenet数据集包含超过1400万张图片[18],自然语言处理中的xlnet使用了高达20亿单词的百科语料库[19]。但很多时候,要获取大量的数据是不可行的[20]。例如,在药物发现中,通常只有很少的分子被选中用于合成新的药物,而在实验室中进行了一系列的实验之后,这些被选中的分子又很可能因为没有想要的属性而不能成为药物,这些导致了药物发现的数据量极其稀少[21-22],即使是几百条实验的数据也非常的珍贵。此时,如果使用机器学习方法直接在少量数据集上进行训练,难以得到一个可靠的分子属性预测模型,机器学习方法在小样本情况下极易失败。
...................................
1.2分子表示学习
使用机器学习方法预测分子属性需要对分子进行表示,即将分子进行数据化,使得其能够被计算机处理,能够用于机器学习方法或模型的训练。在图像领域,图像被表示为多维矩阵,矩阵中每个元素代表图像上的一个像素点,通过卷积网络等深度学习网络将图像从三通道的矩阵表示转成向量表示,将这个向量表示用于后续的分类或回归预测。在自然语言处理领域,首先构建一个词典,然后根据词典大小将词表示为one-hot向量,使得模型能够处理。但分子可以建模为化学式表示的字符序列、可以建模为图结构的形式,对分子的多种建模使得分子不能直接使用图像或自然语言处理的方法进行表示,必须寻找适合的分子表示方法。
文献[23]使用库伦矩阵表示一个分子,库伦矩阵中的非对角元表示两个原子之间的库伦核排斥力(Coulomb repulsion),对角元近似为原子的电势能;文献[24]则提出了一种键袋表示法(Bag of Bond),键袋(Bag of Bonds,BOB)表示法专门使用库伦矩阵的元素,将不同原子对的元素分组到不同的袋子中,并根据它们的相对大小在每个袋子中对它们进行排序;BAML(Bonds,angles,machine learning)分子表示[25]可以看作是BOB的多体扩展,对于键合原子和非键合原子,所有成对的核排斥分别被Morse/Lennard-Jones势所取代,此外,共价键合原子之间的三体和四体相互作用分别使用角度项和扭转项进行计算,参数和函数形式基于通用力场(UFF);扩展连接性指纹(ECFP4)[26]是一种常见的分子表征,分子指纹的基本思想是将一个分子表示为一组固定直径的子图,为了产生一个定长向量,可以对子图进行散列,使每个子图将定长向量中的一位设置为1,ECFP4仅基于指定所有共价键的分子图,例如由SMILES字符串编码的共价键。管这些分子表示方法用于分子属性预测获得了显著的成就[10],但上述的这些分子表示通常需要手动构建分子特征,或者需要额外的计算,或者需要深厚的专业背景知识,这些原因导致上述分子表示方法在实践中受到较多的限制。
................................
第二章 理论知识
2.1 相似度度量
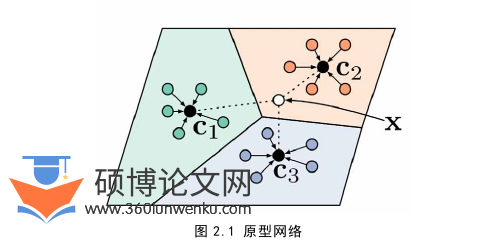
在小样本学习中,相似度度量具有重要意义。例如,匹配网络中使用类似于最近邻的分类思想对未知样本进行分类,原型网络用原型表示一个类,对未知样本,计算其与每个类原型的距离,将其划分到距离最小的那一个类中。距离能够度量相似度,本节介绍本文中所使用的相似度度量。
文献[54]训练了一个“孪生网络”,该网络输入两个样本,使得标签相同的样本的相似度得分更高,标签不同的样本相似度得分更低。网络训练完成之后,对于one-shot任务,输入支持样本和查询样本,根据网络输出的相似度得分判断查询样本是否属于该类。匹配网络[50]使用深度卷积神经网络作为嵌入函数,训练两个特征提取器,一个提取支持样本的特征,另一个提取查询样本的特征,另外,在提取出特征后还进行双向嵌入从而得到支持样本和查询样本在嵌入空间中的表示。该工作首先将数据集划分为多个不同的幕,每一个幕作为一个batch输入到网络中,网络将样本映射到嵌入空间之后,在嵌入空间使用余弦距离作为相似度评判标准,并使用K近邻分类方法对查询样本进行分类。
计算机论文怎么写
............................
2.2图表示学习
在分子属性预测中,分子可以建模为一个图,图中的节点表示分子中的原子,节点之间连接的边表示两个原子之间的化学键。将分子建模为图后,便可以使用图神经网络等图表示学习模型获取分子的隐藏特征表示,将图结构的分子数据转换为向量,用于后续的分类或回归任务。图神经网络是图表示学习的主要方法,另一种有趣的方法是Graphormer。本节将分别介绍图神经网络和Graphormer。
章首先对小样本分子属性预测问题进行了建模。给出一些和目标任务相关的任务,本文希望在相关任务中学习一些知识,将这些知识用于解决目标任务。然后,本章介绍了一些和本文密切相关的经典的小样本学习(元学习)方法,如原型网络,匹配网络,模型无关元学习等。之后,本章介绍了小样本分子属性预测领域使用的一些方法如迭代精炼长短期记忆网络等。另外,本章还介绍了两种用于分子图表示学习的模型——图神经网络和Graphormer,同时,本章也介绍了注意力机制,并从注意力机制最有影响力的工作Transformer中推导出本文第三章所使用的注意力机制。
.............................
第三章 基于注意力编码的小样本分子属性预测 ................................... 18
3.1 基于注意力编码的小样本分子属性预测 ................................... 18
3.2 实验结果与分析 .............................................. 20
第四章 分子表示模型alpha-Graphormer ............................ 30
4.1分子表示模型alpha-Graphormer .............................. 30
4.2 实验结果与分析 ........................................ 32
第五章 小样本分子属性预测的数据集Chembl-2000 ...................... 36
5.1 Chembl-2000数据集 ................................... 36
5.1.1 Chembl数据集分析 ......................................... 36
5.1.2 Chembl-2000数据集构建 ..................................... 36
第五章 小样本分子属性预测的数据集Chembl-2000
5.1Chembl-2000数据集
本文基于Chembl数据集,构建了一个用于小样本分子属性预测的新数据集,本文将其命名为Chembl-2000。本章将通过数据分析、数据集构建、数据集划分、评价机制等方面说明本文所构建的Chembl-2000数据集。除此之外,本文实现了匹配网络[50],原型网络[52],注意力编码的baseline。
5.1.1 Chembl数据集分析
Chembl[68]数据集共包含近500000个分子,1310个化学实验的实验记录。本文从文献[68]中的开源代码库中下载相关数据。
因该数据集中存在一些分子,无法用Rdkit建模,故本文首先将这些分子从整个数据集中剔除,获得了包含456309个分子1310个化学实验的数据集。该数据集中,样本数最多的任务拥有86690个分子的实验数据,样本数量最少的任务仅仅包含了32个分子。如表5-1所示,不超过两百个样本的实验总数共有523个,在任务量中占比最高。超过8000个分子的实验则有134个。

计算机论文参考
...................................
第六章 总结与展望
分子属性预测在药物发现、材料发现等领域有着广阔的前景和重大的意义。但当前基于机器学习特别是深度学习的分子属性预测方法需要提供大量的数据,如果数据量少,要获得一个有效的模型几乎是不可能的,而很多时候要获取大量的数据是不现实的,如何在样本数量极少的情况下获得一个可用的分子属性预测模型是本文工作的重心。图像领域的小样本学习方法揭示了处理这个问题的一条路径,但图像分类和分子属性预测存在很大的区别,无法直接应用图像领域的小样本学习方法处理小样本分子属性预测问题。
本文使用元学习理念对小样本分子属性预测问题进行了建模,本文认为,在这个建模条件下,解决小样本分子属性预测问题有三个关键因素,第一,需要一个强而有力的分子表示学习模型用于学习分子的特征表示;第二,需要一个更好的小样本学习算法才能提高模型对小样本分子属性的预测能力;第三,需要一个任务丰富的高质量数据集,使得模型可以从该数据集中学到和目标任务相关的知识。本文针对这三个关键因素提出了本文的解答方案。
首先,对于图表示学习模型,本文改进了一个有趣且及其强大的图表示学习模型——Graphormer。本文通过实验揭示了Graphormer中图结构信息的表现比自注意力更加好这一现象,依据这一现象,本文提出Graphormer中图结构信息比自注意力更加重要这一假设,基于该假设,本文引入了一个缩放因子,缩放因子能够引导模型对自注意力和图结构信息赋予不同的关注度,本文将改进的方法称为alpha-Graphormer。本文在Tox21数据集上的实验表明,本文改进的方法alpha-Graphormer在Tox21数据集上具有比原模型Graphormer更好的表现。
其次,对于更好的小样本学习方法,本文提出了注意力编码。注意力编码的关键想法来源于人类的生活,人类在认识新事物时,通常将其与旧事物相联系,从而加深对新事物的认识。基于该想法,本文提出了一种基于注意力的编码方法,该方法能够建立已知分子和未知分子之间的联系。本文提出的注意力编码方法首先将支持分子和查询分子输入特征提取器得到分子的嵌入特征表示,然后分子的嵌入特征表示通过注意力编码获得最终表示,之后使用基于注意力机制的K-近邻方法对未知分子进行分类。本文在Tox21、Sider、Chembl20数据集上的实验表明,注意力编码方法能够提高模型的预测性能,并且,注意力编码方法获得了超过匹配网络,全条件嵌入,原型网络的性能,获得了与有属性意识的关系网络有竞争力的性能,但由于本文未使用自监督预训练,所以本文的注意力编码性能表现不如使用了自监督预训练的方法。
参考文献(略)