1 绪论
1.1 研究背景及意义
在人类基因组中,DNA 的转录不仅存在于蛋白质编码区,而是存在于包括非编码区域的整个基因组中。非编码 DNA 的比例占总基因组 DNA 的近 98.5%,这就使得 DNA转录形成的 RNA 绝大多数为非编码 RNA(Non-coding RNA,ncRNA)(Birney E,2017)。由于在广泛的生物过程中的重要作用,非编码 RNA 的研究代表了当前生物医学研究中最令人兴奋的领域之一(Liang C et al.,2017;Jiang L et al.,2020)。通常,非编码 RNA分为小部分大于 200 个核苷酸(nt)的长非编码 RNA(Long Non-coding RNA,lncRNA)和大多数的 20 到 35 个核苷酸的小非编码 RNA(Small Non-coding RNA,sncRNA)(PerkelJ M,2013;Mori T et al.,2018)。
1.1 研究背景及意义
在人类基因组中,DNA 的转录不仅存在于蛋白质编码区,而是存在于包括非编码区域的整个基因组中。非编码 DNA 的比例占总基因组 DNA 的近 98.5%,这就使得 DNA转录形成的 RNA 绝大多数为非编码 RNA(Non-coding RNA,ncRNA)(Birney E,2017)。由于在广泛的生物过程中的重要作用,非编码 RNA 的研究代表了当前生物医学研究中最令人兴奋的领域之一(Liang C et al.,2017;Jiang L et al.,2020)。通常,非编码 RNA分为小部分大于 200 个核苷酸(nt)的长非编码 RNA(Long Non-coding RNA,lncRNA)和大多数的 20 到 35 个核苷酸的小非编码 RNA(Small Non-coding RNA,sncRNA)(PerkelJ M,2013;Mori T et al.,2018)。
其中,长非编码 RNA 在生物学中的功能机制通常和疾病相连(Lina M et al.,2019)。例如,PCGEM1 是在前列腺肿瘤中两个过度表达的非编码 RNA 之一,其参与调节细胞的生长,与增殖和集落形成增加有关(Kang Y et al.,2016)。还有,MALAT1 是最初被鉴定为在早期非小细胞肺癌转移过程中上调的高表达的非编码 RNA(Yoshimoto R etal.,2016),在多种生理过程中得到鉴定,例如选择性剪接,核组织,基因表达的表观遗传调控(Wu Y et al.,2015),并且更多证据表明 MALAT1 也与各种病理过程密切相关(陈炜等,2019),从糖尿病并发症到癌症,其过表达程度是患者存活率的早期预后指标(Yoshimoto R et al.,2016;Liu J H et al.,2014)等等。
小非编码 RNA 发挥着基因表达调控的作用,主要包括的三大类:小分子 RNA(microRNA,miRNA)、小分子干扰RNA(siRNA)和与piwi蛋白相作用的RNA(piRNA)(Rigoutsos I et al.,2019)。在小非编码 RNA 中,miRNA 和 siRNA 的长度通常为 21nt左右,并且已经进行了广泛的研究。而起步较晚的 piRNA 的长度集中在 24nt 至 34nt 之间,存在于至少数十万种多细胞生物中,该数目远远超过其他类型的已知 RNA 的总数(Ku H Y et al.,2014)。
............................
小非编码 RNA 发挥着基因表达调控的作用,主要包括的三大类:小分子 RNA(microRNA,miRNA)、小分子干扰RNA(siRNA)和与piwi蛋白相作用的RNA(piRNA)(Rigoutsos I et al.,2019)。在小非编码 RNA 中,miRNA 和 siRNA 的长度通常为 21nt左右,并且已经进行了广泛的研究。而起步较晚的 piRNA 的长度集中在 24nt 至 34nt 之间,存在于至少数十万种多细胞生物中,该数目远远超过其他类型的已知 RNA 的总数(Ku H Y et al.,2014)。
............................
1.2 国内外研究现状
由于非编码 RNA 在细胞过程和疾病发展中的重要作用,众多研究者用生物实验和生物信息学方法来识别预测非编码 RNA 及其功能。在生物实验方法上,酶法和化学法RNA 测序、专用 c DNA 文库并行克隆 ncRNAs、微阵列分析和基因组 SELEX 是最常用的方法(常朝霞,2019)。然而,实验方法昂贵且耗时,因此也开发了数百种计算方法,以优先考虑高度置信的 ncRNA 候选,以进行进一步的实验验证(Zhang Y et al.,2017)。
由于非编码 RNA 在细胞过程和疾病发展中的重要作用,众多研究者用生物实验和生物信息学方法来识别预测非编码 RNA 及其功能。在生物实验方法上,酶法和化学法RNA 测序、专用 c DNA 文库并行克隆 ncRNAs、微阵列分析和基因组 SELEX 是最常用的方法(常朝霞,2019)。然而,实验方法昂贵且耗时,因此也开发了数百种计算方法,以优先考虑高度置信的 ncRNA 候选,以进行进一步的实验验证(Zhang Y et al.,2017)。
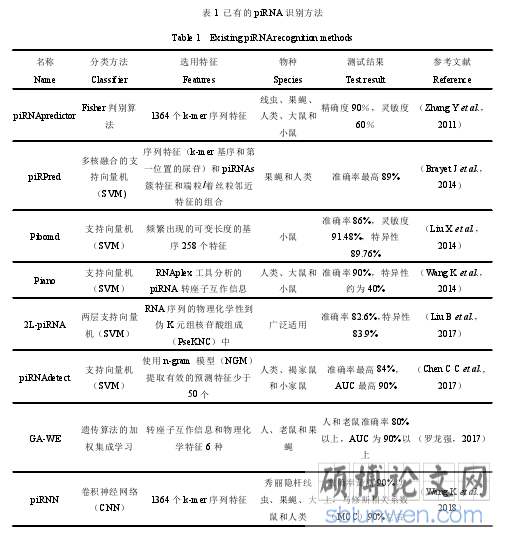
高通量测序技术的快速发展,使得生物数据的获取更加容易,从大数据量的测序结果中寻找和挖掘有价值的信息是个极具潜力的挑战,生物大数据已经成为生命科学研究领域创新的源泉,农业生产的研究和发展同样离不开生物科学的发展,利用生物信息学相关工具就能解决在农业上对农作物品种改良和丰富种质资源等问题,因此,将生物信息学的相关计算机技术应用到农业生产发展中对我国现代化发展进程具有重要的意义(徐佳莹等,2019)。越来越多的研究试图通过生物信息学的方式来解决序列的分析和识别问题(王媛媛,2019;程功,2015),其中包括 piRNA 的识别,其中目前已有的算法按时间综述如表 1 所示。
..............................
2 piRNA 来源和识别方法
2.1 非编码 RNA 概述
非编码 RNA(ncRNA)是一种从 DNA 中转录但不编码蛋白质的功能性 RNA。根据转录组学和生物信息学的研究,有成千上万的 nc RNA 根据其功能和长度被分为不同的类别,包括 tRNA、r RNA、miRNA、siRNA、piRNA 和 lncRNA 等,还包括很多未知功能的 RNA(Chen L et al.,2017)。
这些 ncRNA 在各种细胞过程中发挥着重要作用。例如,r RNA 在翻译过程中催化氨基酸之间的肽键形;miRNA 在转录过程中很重要,并对基因表达进行转录后调节;lncRNA 在 X 失活、印记、表观遗传标记和基因表达的调节中起着关键的多种作用,此外,它们在各种疾病中也显示出极大的重要性(Zhang Y et al.,2017)。目前,仍然只有极少数的非编码 RNA 通过生物实验证明具有明确的生物学意义,大量的非编码 RNA的功能依然未知(李建伟等,2017)。
2.1 非编码 RNA 概述
非编码 RNA(ncRNA)是一种从 DNA 中转录但不编码蛋白质的功能性 RNA。根据转录组学和生物信息学的研究,有成千上万的 nc RNA 根据其功能和长度被分为不同的类别,包括 tRNA、r RNA、miRNA、siRNA、piRNA 和 lncRNA 等,还包括很多未知功能的 RNA(Chen L et al.,2017)。
这些 ncRNA 在各种细胞过程中发挥着重要作用。例如,r RNA 在翻译过程中催化氨基酸之间的肽键形;miRNA 在转录过程中很重要,并对基因表达进行转录后调节;lncRNA 在 X 失活、印记、表观遗传标记和基因表达的调节中起着关键的多种作用,此外,它们在各种疾病中也显示出极大的重要性(Zhang Y et al.,2017)。目前,仍然只有极少数的非编码 RNA 通过生物实验证明具有明确的生物学意义,大量的非编码 RNA的功能依然未知(李建伟等,2017)。
随着高通量技术在不同的细胞和组织中的广泛使用,已鉴定的非编码 RNA 的数量也急剧增加。在 ENCODE 版本 29 中已经注释了人类基因组中的 4 万多个非编码 RNA。非编码 RNA 会影响癌症的发生、发展和结果(Liu J H et al.,2014),许多非编码 RNA是十分重要的癌基因和抑癌基因,例如,由于表观遗传失调沉默 miRNA-127 从而增强了胃癌中 MEK1 和 TEAD4 的表达(郑涵等,2019);lncRNA HULC 通过降低 ATG7和 ITGB1 在上皮性卵巢癌中作为癌基因(Chen S et al.,2017),以及 lncRNA HOXB-AS3编码的肽抑制结肠癌的生长(Huang J Z et al.,2017)。由于其广泛且重要的作用,近几十年来,非编码 RNA 研究的逐渐成为热点(Jiang L et al.,2020)。
........................
2.2 piRNA 概述
与 Piwi 蛋白相作用的 RNA(Piwi-interactingRNA,piRNA)是在 2006 年发现的一类复杂的小非编码 RNA,长度约为 24 到 34 个核苷酸,其能够与 Piwi 亚家族中 Argonaute蛋白质相作用(Lau et al.,2006;Brayet J et al.,2014)。
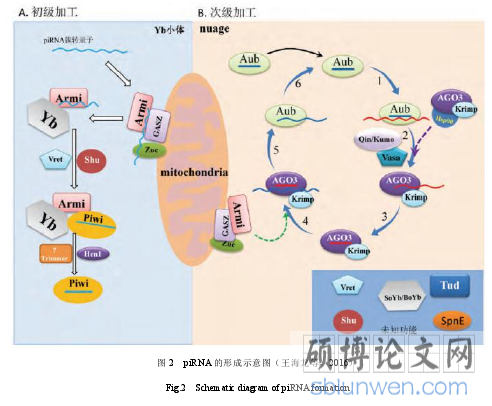
piRNA 的成熟有两个加工过程,如图 2 所示(王海龙等,2016),piRNA 的生成及其功能依赖于 PIWI 蛋白,piRNA 序列来源于基因组的转座子、编码基因区和特异的基因间位点。piRNA 作为 PIWI 的向导调控靶基因的表达以及转录和转录后水平的修饰。在果蝇卵巢生殖细胞中,piRNA 簇的转录子转运到细胞的胞浆后经过初级加工的途径形成初级 piRNA,能够结合到 Piwi 和 Aub 上,生殖细胞的初级 piRNA 还能够进入到次级加工途径,经过 PIWI 家族蛋白的协同及加工,能够使细胞中的 piRNA 大量扩增,称为乒乓循环,但是目前对乒乓循环的细节和具体机制还所知甚少(王海龙等,2016)。
与 Piwi 蛋白相作用的 RNA(Piwi-interactingRNA,piRNA)是在 2006 年发现的一类复杂的小非编码 RNA,长度约为 24 到 34 个核苷酸,其能够与 Piwi 亚家族中 Argonaute蛋白质相作用(Lau et al.,2006;Brayet J et al.,2014)。
piRNA 的成熟有两个加工过程,如图 2 所示(王海龙等,2016),piRNA 的生成及其功能依赖于 PIWI 蛋白,piRNA 序列来源于基因组的转座子、编码基因区和特异的基因间位点。piRNA 作为 PIWI 的向导调控靶基因的表达以及转录和转录后水平的修饰。在果蝇卵巢生殖细胞中,piRNA 簇的转录子转运到细胞的胞浆后经过初级加工的途径形成初级 piRNA,能够结合到 Piwi 和 Aub 上,生殖细胞的初级 piRNA 还能够进入到次级加工途径,经过 PIWI 家族蛋白的协同及加工,能够使细胞中的 piRNA 大量扩增,称为乒乓循环,但是目前对乒乓循环的细节和具体机制还所知甚少(王海龙等,2016)。
.........................
3.1 原始数据获取.................................16
3.1.1 正数据获取......................................16
3.1.2 负数据获取...............................17
4 基于深度学习的 piRNA 识别算法...............................24
4.1 DeepiRNA 模型设计..............................24
4.1.1 CNN 阶段............................... 26
4.1.2 BiLSTM 阶段........................... 27
5 DeepiRNA 系统设计与实现......................... 40
5.1 系统设计....................................43
5 DeepiRNA 系统设计与实现
5.1 系统设计
DeepiRNA 系统后端算法使用 Python 语言,结合 Web 前端微服务框架进行开发,使用 Docker 应用容器引擎将后端部署到腾讯云轻量级服务器上,实现系统的前后端分离。服务器操作系统为 64 位 CentOS,版本为 7.6。
DeepiRNA 系统后端算法使用 Python 语言,结合 Web 前端微服务框架进行开发,使用 Docker 应用容器引擎将后端部署到腾讯云轻量级服务器上,实现系统的前后端分离。服务器操作系统为 64 位 CentOS,版本为 7.6。

DeepiRNA 系统设计如图 32 所示,系统实现一共实现四个模块,在网站上分别显示为四个页面,包括主页模块、模型介绍模块、在线预测模块和通讯模块。
主页模块实现系统首页功能;模块介绍模块介绍了 DeepiRNA 模型设计的目的和意义,并且展示了 DeepiRNA 的模型结构;在线预测模块是系统的功能模块,实现用户预测自己的序列的目的;通讯模块对本实验所属的实验室做了简介,本文中不再详述。
............................
6 总结与展望
6.1 总结
随着小非编码 RNA 的研究逐渐深入,piRNA 也被越来越多的研究人员所熟知,piRNA 的数据也逐渐丰富起来。想比于用生物实验方式验证 piRNA 序列的长周期处理复杂的特点,使用计算的方法来识别 piRNA 是一种更为省时的提供给生物专家的初步筛选方式,对进一步生物研究具有重要价值和意义。本文对当前 piRNA 序列计算识别方法的国内外发展现状以及发展趋势做了大量的研究,通过对比分析的方式总结概述了不同的文章中采用方法的优缺点,就目前的前人研究工作,仍有许多待提高的部分。本文针对以上问题,做的主要工作如下:
(1)分析了解决基于深度学习的 piRNA 识别算法的具体思路。
通过分析总结之前的研究,提出本文的主要研究内容,即减少人工提取特征,使用深度学习的方式从非编码 RNA 中识别 piRNA 序列。首先,对原始数据从实用性和进一步研究的角度进行分析,选择小鼠和人类为本文研究物种,并且介绍了相关使用到的数据库,包括 piRBase 数据库和 MiRBase、GtRNAdb 等原始负数据库。然后,通过分析选择深度学习算法,能够解决大量数据的特征选取和序列识别的问题,最终选择了卷积神经网络(CNN)和双向长短时记忆网络(BiLSTM),并对两种算法的特点和使用到的方法以及算法原理做了详细的描述。
6.1 总结
随着小非编码 RNA 的研究逐渐深入,piRNA 也被越来越多的研究人员所熟知,piRNA 的数据也逐渐丰富起来。想比于用生物实验方式验证 piRNA 序列的长周期处理复杂的特点,使用计算的方法来识别 piRNA 是一种更为省时的提供给生物专家的初步筛选方式,对进一步生物研究具有重要价值和意义。本文对当前 piRNA 序列计算识别方法的国内外发展现状以及发展趋势做了大量的研究,通过对比分析的方式总结概述了不同的文章中采用方法的优缺点,就目前的前人研究工作,仍有许多待提高的部分。本文针对以上问题,做的主要工作如下:
(1)分析了解决基于深度学习的 piRNA 识别算法的具体思路。
通过分析总结之前的研究,提出本文的主要研究内容,即减少人工提取特征,使用深度学习的方式从非编码 RNA 中识别 piRNA 序列。首先,对原始数据从实用性和进一步研究的角度进行分析,选择小鼠和人类为本文研究物种,并且介绍了相关使用到的数据库,包括 piRBase 数据库和 MiRBase、GtRNAdb 等原始负数据库。然后,通过分析选择深度学习算法,能够解决大量数据的特征选取和序列识别的问题,最终选择了卷积神经网络(CNN)和双向长短时记忆网络(BiLSTM),并对两种算法的特点和使用到的方法以及算法原理做了详细的描述。
(2)进行了正负数据准备,做了数据分析与预处理工作。
准备人类和小鼠实验数据,包括正数据 piRNA 数据和非 piRNA 的三个来源的负数据。负数据包括使用了 piRNN 算法中的一阶马尔科夫模型生成的相应仿真 piRNA 序列;MiRBase 中下载相应物种的成熟 miRNA 序列;以及从基因组 tRNA 数据库 GtRNAdb数据库中下载 hg38 版本的人类和 mm10 版本的小鼠 tRNA 数据,从序列 5’端开始 0-20随机位置起始随机切割 24nt 到 34nt 之间随机长度的 tRNA 片段。
参考文献(略)
准备人类和小鼠实验数据,包括正数据 piRNA 数据和非 piRNA 的三个来源的负数据。负数据包括使用了 piRNN 算法中的一阶马尔科夫模型生成的相应仿真 piRNA 序列;MiRBase 中下载相应物种的成熟 miRNA 序列;以及从基因组 tRNA 数据库 GtRNAdb数据库中下载 hg38 版本的人类和 mm10 版本的小鼠 tRNA 数据,从序列 5’端开始 0-20随机位置起始随机切割 24nt 到 34nt 之间随机长度的 tRNA 片段。
参考文献(略)