本文是一篇计算机论文,本文在数据库和论文的公开数据集上进行benchmark实验,通过对比公共指标展现出来AntDPP模型出色的性能。并针对MET和SARS-V2抗原进行了下游实验,并通过广泛的对比实验、消融实验和超参数敏感性实验,并通过湿实验的方式验证工作流的有效性。
第1章 绪论
1.1 研究背景及意义
在人类疾病的治疗中,抗体发挥着至关重要的作用[1, 2]。抗体是一类重要的蛋白药物[3, 4],是免疫系统对抗原产生的蛋白质,其具有中和或清除体内靶向抗原的能力,对于抵抗感染和疾病至关重要。为了挖掘抗体治疗的巨大潜力,必须采用更合适的技术来合理设计抗体[5]。传统的抗体设计方法,如杂交瘤、噬菌体展示技术、定向进化和诱变技术等[6, 7],通常使用人工定义的能量函数设计抗体[8-10],但此类方法需要大量的计算资源,且由于链间相互作用的复杂性无法获取确定性的力场结构[11-14],无法满足快速和精确设计抗体的需求。近年来随着人工智能(Artificial Intelligence,AI)的快速发展,将AI与生物制药技术相结合来寻找特效药成为一条可靠的研究路径[15-21]。
目前基于深度学习方法的抗体序列设计方法已经取得了一系列的研究成果。与人类自然语言是由字母组合相类似,抗体序列是由二十种氨基酸组成,通过不同组合排列形成不同的功能,因此大量的研究将自然语言处理的深度学习方法用于抗体设计。如LM-DESIGN[22]、ESM[23]和RefineGNN[24]等方法尝试将自然语言模型迁移到抗体序列设计上,通过预训练模型得到更好的抗体表示,用于下游任务如抗体功能分类、抗体与特定抗原的亲和力、结合力、蛋白性质等取得了较好的结果[25-27]。
............................
1.2 国内外研究进展
1.2.1 基于序列蛋白预训练国内外研究现状
在不考虑抗原-抗体是否配对的情况下,序列数据则更容易获取[28]。判断抗原抗体是否具有高亲和力。抗体和抗原如何有效编码则作为首要的问题[23, 38]。抗原抗体的序列数据是由二十个氨基酸的首字母组成,其内在联系与自然语言类似因此当前有很多工作将自然语言的工作进行迁移如ProtTrans[39],ESM[23],antiberty[26],antiberta[40]和BERTTransformer[27]等。这些工作为抗原抗体编码做下游任务时都是十分必要。目前绝大多数抗原抗体结合(Antigen Antibody Interaction, AAI)工作优先考虑将两个编码链接在一起通过Dense层后判断是否结合,能够获得比传统机器学习更好的结果。但此类方法在遇到全新的抗原抗体对的时候模型表现力较为有限,鲁棒性差。
特异性是决定抗体与抗原结合的重要因素,面向特异性设计的需求,一些研究将小分子生成用在抗体设计,如以GAN为架构的生成模型如ORGAN[41],SeqGAN[42]。但是其存在离散数据无法微分的问题,序列长度偏短,训练困难,可复线能力差。自回归模型在少量数据时往往无法满足多样性需求,生成的序列更像是已有数据的排列组合[43]。早期分子生成也有类似的工作Junction Tree[44],通过对序列化学或物理性质构建成树方式生成模型做出不符合自然规律的动作。强化学习中判别器,会影响生成模型的偏好,因此判别器的设计就显得尤为重要。此前作为判别器的Absolute[31]其判断原理是通过两个蛋白之间三维结构的距离和形状来判断其是否能够结合,通过分子动力学的方式进行模拟,需要消耗大量时间和资源,在实际验证中其准确率也有待考量。抗体设计存在一些生成规则,如电荷能量不能过高,相同氨基酸不能连续超过四个等[24]。由于这些 Y 形蛋白质的结合特异性在很大程度上取决于它们的互补决定区(CDR),因此计算抗体设计的主要目标是自动创建具有所需特性的CDR子序列。大多数生成的时候只会考虑CDR区域而不考虑全长序列的生成[24, 33]。然而不同区域的CDR长度和氨基酸的分布有所不同,需要在设计时单独考虑。
......................
第2章 相关理论知识与定义
2.1 抗体&抗原相关背景知识
计算机论文怎么写
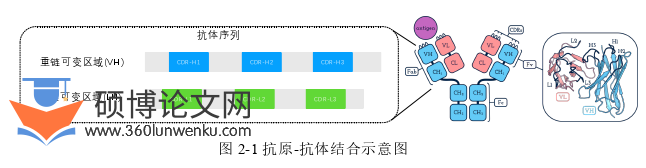
抗体能够与抗原(如病毒)结合,并将其呈递给免疫系统,从而激发免疫反应。中和抗体这一亚群不仅能与抗原结合,还能抑制其活性。抗体由重链和轻链组成,每个链包含一个可变区(VH/VL)和若干恒定区。重链可变区又细分为框架区和三个互补确定区(CDR)。重链上的三个CDR被标记为CDR-H1、CDR-H2和CDR-H3,每个CDR对应一个连续的子序列如图2-1。灰色区域作为框架区域(Framework Regions,FR)是抗体分子中链接CDRs的结构支架,通常具有较高的保守性和较低的异变性,其主要作用是维持抗体分子的整体结构稳定和凝聚力。对应蓝色和绿色作为抗体的重链的互补决定区域(Complementarity-Determining Regions,CDRs)和轻链的互补决定区域,CDRs是抗体分子上特异性结合抗原的部分,分为CDR1、CDR2和CDR3。CDRs通过与抗原相互作用,决定了抗体的抗原结合特异性和亲和力,因此在抗体识别和中和病原过程中起着关键的作用。在图中可以看出在CDRs和框架区域之间存在一个比较短的区域被称为互补区(Complementarity Regions,CRs),其具有相对较高的变异性和较低的保守性。互补区在抗体与抗原结合时提供了柔性和多样性,有助于适应各种抗原结构。可变最高的部分CDRs是决定结合和中和的关键[64]。本文将抗体设计形式化为根据框架区生成CDR的任务,模型将抗体的序列、对应区域信息、氨基酸性质和三维结构有机融合,符合生物学逻辑[65, 66]。使用生物信息作为提示信息可以在优化抗体序列时更好地了解抗体结构中各个区域的功能和作用,可以指导模型用更少的参数和训练时间发现具有特定功能和性质(如增强抗原结合亲和力,减少免疫原性等)的抗体分子。综合考虑抗体区域、性质和结构有助于预测抗体与抗原之间的相互作用模式和结合位点,从而增强模型的鲁棒性。本文提出了一种将三维结构与序列结合的自回归模型,并根据固定框架区进行条件生成。
.....................
2.2 预训练模型相关背景知识
预训练模型是在大型数据集上训练得到的机器学习(ML)模型,可进一步微调以适用于特定任务。通常,预训练模型被用作开发ML模型的起点,因为它们提供了一组初始的权重和偏置,可供针对特定任务进行微调。
使用预训练模型有多个优势,包括能够借鉴他人的知识和经验、节省时间和资源,以及提高模型性能的能力。这些模型通常在大型且多样化的数据集上进行训练,已经学会识别各种模式和特征。因此,它们为模型微调提供了坚实的基础,并且可以显著提高模型的性能。
预训练模型有多种类型,例如语言模型、目标检测模型和图像分类模型。卷积神经网络通常用作图像分类模型的基础,这些模型被训练用于将图像分类到预定义的类别中(CNNs)。
另外,卷积神经网络或基于区域的卷积神经网络通常用作目标识别模型的基础,这些模型被训练用于在照片或视频中识别和分类物体(R-CNNs)。而循环神经网络(RNNs)或变压器通常被用作语言模型的基础,这些模型被训练用于预测序列中的下一个单词。
总的来说,预训练模型是ML中一种强大的工具,可作为开发ML模型的起点。它们提供了一组初始的权重和偏置,可供特定任务的微调,并且可以显著提高模型的性能。
生物预训练模型是一种机器学习模型,它是在生物数据(如DNA序列、蛋白质序列或分子结构)上进行训练的,而不是像文本或图像这样的通用数据集。这些模型专门设计用于捕获生物数据中存在的模式和特征,可以用于生物信息学、计算生物学和相关领域的各种任务。生物预训练模型可以使用不同的架构和技术进行训练,具体取决于手头的特定任务。例如,在生物信息学中,可以使用循环神经网络(RNNs)或变压器进行序列预测或生成任务,而卷积神经网络(CNNs)可以用于生物图像的分析任务。
..............................
第3章 基于多维度生物信息的预训练生成模型 ...................... 16
3.1 问题定义与方法描述 .......................... 16
3.2 基于特定互补区域信息提取 ...................... 17
3.3 抗体结构信息构建 ......................... 18
第4章 基于虚拟筛选和微调的抗体序列优化 ........................ 27
4.1 问题定义和方法描述 ....................................... 27
4.2 虚拟筛选器定义 ........................................... 28
第5章 实验与分析 ................... 39
5.1 实验环境介绍 ......................... 39
5.2 实验前期准备 ............................. 39
第5章 实验与分析
5.2 实验前期准备
在本文中,本文采用了用SAbDab这一类数据库,将大部分通过一定规则的方式进行筛选后得到较高质量的数据用来做预训练模型,后在较新的数据上进行优化序列并做出一些计算机模拟的验证对比。
5.2.1 实验数据集
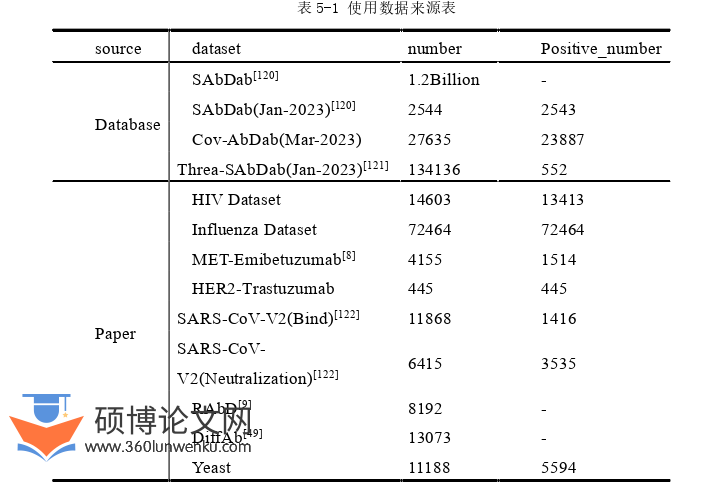
为了防止预训练的数据出现在下游任务中,本文首先进行去重的操作。为了获得较高质量的数据,我们首先进行了对数据进行清洗,如图5-1所示。其中不同数据包含了不同的类型的抗体。
OAS(The Observed Antibody Space)是一个综合性数据库(包含表格中四个数据集),包含超过10亿条抗体可变区序列,这些序列来源于80项免疫库鉴定研究。该数据库涵盖了来自人类、小鼠、大鼠、骆驼、兔子和食蟹猴等6种不同物种的抗体重链和轻链。 为了开始研究,先分别排除了具有相同成分的重链和轻链序列。接着,对于重链和轻链,识别出具有重复互补决定区3(CDR3)的剩余序列,并使用LinClust算法[119]根据整个序列的95%同一性阈值对它们进行聚类。然后,选定的序列按照80:10:10的分配比例分割成非重叠的训练、验证和测试集。训练集总共包含201,845,244条重链和16,492,030条轻链序列,而另外保留了25,230,656条重链和2,061,504条轻链序列的两个评估集,以供未来评估、超参数调优和遮蔽氨基酸恢复测试。
计算机论文参考
............................
第6章 总结与展望
本论文针对抗体序列的生成和优化问题,提出了一种基于多维度的抗体预训练模型。在现有抗体蛋白结构和序列自监督模型的基础上,从构建合理的氨基酸信息表示,正确聚合抗体结构和信息和抗原-抗体结合强度的表示,提出了基于多维度和预训练的自监督抗体序列优化模型。本文主要工作和贡献如下:
提出基于多尺度和预训练的抗体序列生成模型,该模型是基于抗体先验生物先验信息和结构信息预训练而来,克服了传统方法缺少多维度抗体特征信息的问题。本文基于Chothia方法得到氨基酸区域位置信息,通过生物学家总结氨基酸性质和对应序列编号,并通过表示学习方式进行嵌入表示,之后通过聚合的方式将三种属性整合为不同位置相同或不同氨基酸进行隐式表示。对于抗体三维结构,能获取到的数据极为有限,因此本文采取了在蛋白结构预测的预训练模型进行微调。使用Graph Transformer的网络架构进行获取结构特征的嵌入表示。最后将氨基酸的序列表示与结构进行对齐。此外,构建了基于氨基酸属性和三维结构的预训练模型,使模型学习到抗体设计的规则,在满足设计规则的条件下尽可能的探索抗体序列数据分布空间。
提出基于多尺度和预训练的抗体序列优化,在较为广阔的抗体序列数据分布空间中针对特定抗原尽可能搜索更符合某些特征(如亲和力、结合力)的抗体序列数据分布。提出通过Lora的方式进行微调模型和基于虚拟筛选器的微调。虚拟筛选器基于预训练和序列分块聚合的方式设计,解决了注意力机制在长序列上的稀疏问题。对于抗体则采用本文预训练模型的输出。在预测抗体-抗原结合强度时将两个空间分布联系在一起进行微调。
参考文献(略)