本文是一篇药学论文,本研究使用两种生物信息学分析方法(WGCNA分析和基于LASSO算法预后模型的构建分析)筛选出8个食管癌核心基因,并构建预测模型评价预后预测价值,结合临床预后数据最终确定ESCA的不良预后与KIF4A、CDKN3、CDT1、RRM2和MYBL2的过表达以及CELA2A、PGA3和PGA4的低表达有关。
第一章前言
1.1食管癌概述
药学论文怎么写
食管癌(Esophageal Cancer,ESCA)是对中国居民生命伤害严重和高发的恶性肿瘤之一,并且造成的疾病负担严重。吸烟、饮酒、饮食结构和方式不合理和遗传等诸多因素都会导致ESCA的发生和发展概率增大[1]。ESCA包含食管腺癌(EAC)和食管鳞状细胞癌(ESCC)。ESCA患者人数的增加起了医学界的广泛关注,发病率也在稳步上升[2]。食管癌具有复杂的分子机制,通过下调Bax、上调Bcl-2、Bcl-xl和Survivin、p53突变和Fas表达变化来逃避细胞凋亡[3]。尽管ESCA的治疗方法很多,如手术、化疗等,并取得了很大进展,但预后仍然较差[4]。当前临床治疗ESCA最有效的手段还是食管切除术。但是这种治疗方案与许多增加心肌梗死风险的术后并发症有关[5]。
...............................
1.2生物信息学概述
生物信息学是研究生物信息的采集、处理、存储、传播,分析和解释等各方面的学科,也是随着生命科学和计算机科学的迅猛发展,生命科学和计算机科学相结合形成的一门新学科。随着相关高新技术的快速迭代,生物信息学发生了更深层次的变化,使我们能够深入的研究疾病的发生和发展。因此,生物信息学方法的综合应用可以揭示食管癌发生发展的机制,选择出合适的生物标志物,帮助临床医生诊治患者。同时,这也促进我们寻找最佳方案以促进早期发现和改善治疗。生物标志物是反映疾病相关病理性特征的一种重要指标,通常情况下主要用来识别、诊疗和预防疾病,并对预后效果进行分析评价[6]。随着医学技术的日新月异,高通数据量呈现出逐年递增的趋势,主要包括基因组学和蛋白组学等相关数据。在此背景下出现了TCGA、GEO等多组学数据存储平台,这为肿瘤等重大疾病的临床诊疗、预防和预后等创造了有利条件。其中基因组学实现了从机体组织和生物分子层面分析疾病状态下遗传因子发生的具体变化,并筛选出和疾病发生发展及患者预后高度相关的生物标志物。与此同时,诸多医学研究在肿瘤疾病发生发展和生物标志物筛选等方面取得了一定的成果,并已经成功运用到临床实践中[7-10]。梳理相关研究成果发现,借助于高通量数据开展ESCA相关研究的成果资料相对较少,而对于相关生物标志物的研究更是屈指可数。但是临床试验发现生物标志物这种方式存在研究投入成本高和研究效率低下等诸多问题,所以很有必要提前研究潜在的预后生物标志物,然后通过临床试验加以证实。
............................
第二章基于WGCNA食管癌基因的鉴定和分析
2.1数据来源和预处理
选取癌症基因组图谱(TCGA)数据库中的相关数据,主要包括多种癌症相关的临床和多组学数据,并且对于现场研究人员来说是一个非常重要的数据来源。TCGA数据库结合了全球范围内多个种族的34种癌症的肿瘤组织样本及与其相匹配的正常组织。其还包含了一些重要的临床数据,如患者性别、年龄、种族、吸烟状况、癌症阶段、分子亚型和疾病的组织学。它还提供基因组表征的分子数据和信息,如mRNA和蛋白质表达等。研究人员可以从在线TCGA数据库中免费下载目标数据,并对其进行整合和分析,以进一步研究感兴趣的肿瘤的特征。TCGA集成数据集在癌症研究中的使用在世界范围内得到广泛应用,尤其是对泛癌症研究具有深远的影响,因为TCGA等数据库通常包含某些癌症的大量患者样本,并且通常认为对此类大数据的研究可能有助于减少数据偏倚。最近,TCGA数据通常被研究人员用来对肿瘤和正常组织中存在显著表达差异性的基因进行精准识别和筛选,以及这些基因与临床特征的关系。数据集的搜索和搜索过程主要遵循以下具体原则:(1)样本源于人体细胞;(2)样本基因数据源于肿瘤组织及相应正常组织;(3)数据集来自公共资源。
患者的RNA测序数据和临床信息于2022年7月22日从TCGA食管癌队列中下载,该队列可在位于加利福尼亚大学圣克鲁兹分校(http://xena.ucsc.edu/)的Xena网站上获取。在执行WGCNA分析之前,对下载的矩阵信息进行了预处理:从TCGA数据库中的RNA-seq确定其遗传序列是一种ENSG开头的数字序列,即基因Ensembl ID,同时还需得到基因名称Symbol,这样就能更好的掌握后续研究的相关情况。BioMart是一个数据联合框架,其能够为全球范围内的多种数据源提供统一的用户界面[41]。其可以继承现有的多种数据源,并以反向星型模式[42]表示,这样就能显著提升数据检索的效率。许多实验室已成功使用BioMart软件包创建与癌症[43,44]、微阵列[45]和基因表达数据[46]相关集成门户。由此可见,借助于R软件连接Ensembl数据库,并对Ensembl ID进行注释,获取基因Symbol。在经过相应的ID转换后,将相关基因表达量进行log2处理,并将其最为最终表达量。最终得到行为探针,列为基因的矩阵,为构建共表达网络奠定基础。
.................................
2.2研究方法
2.2.1差异基因表达分析
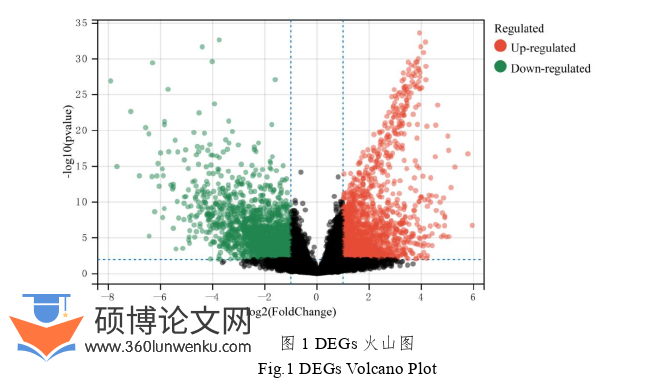
为更加深入的分析研究肿瘤和正常组织样本中基因表达水平的差异性,我们需要先找到与疾病相关的差异表达基因(differentially expressed genes,DEG)。本文使用的是差异基因表达分析。RNA-Seq统计数据通常以表格形式呈现,包括每个样本中不同基因序列片段的数量。需要注意的是相比条件内的变化而言,条件之间变化的量化分析和统计预测尤为重要。因为基因测序数据属于负二项分布,并且不同测序批次的总深度不同,因此本研究利用R Studio软件中的“Limma”功能来识别ESCA与正常食管组织之间的DEGs[47]。同时,计算了食管癌和邻近组织中基因表达水平的倍数变化(FC)。本研究中使用的筛选标准:|log2FC|>1和P<0.01。
在人体患有疾病的初期,人体内的绝大多数基因不会发生改变或者只会出现一些细微的改变,但与此同时可能会有极少数基因会发生显著改变。本研究将借助于火山图和热图对多种基因的变化进行可视化观察分析。火山图Volcano Plot用于显示基因组的差异分布情况,即DEGs表达及分布;而热图则是通过色块色阶对基因的差异表达进行描述,并分析样本间的关系。每个色阶表示对应样本基因表达水平的离散情况。DEG的火山图是用R中的“ggplot2”来进行绘制的。
.......................................
第三章基于LASSO算法食管癌预后模型的构建和验证.............................23
3.1研究对象...............................23
3.2研究方法.....................................24
第四章总结与展望.............................32
第三章基于LASSO算法食管癌预后模型的构建和验证
3.1研究对象
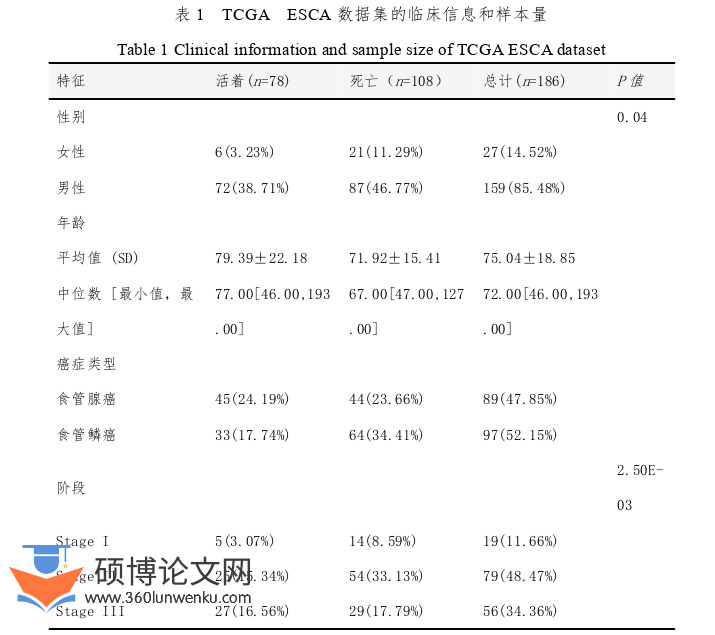
利用R软件对相关数据进行整合处理,对具有一定匹配度的基因表达和临床数据进行整合,进而获取来自170名ESCA患者的160个肿瘤组织和10个正常组织样本(去除16个不符合样本)。样本数据主要包括患者性别、年龄、身高、体重、以及病理分期。RNA测序数据对应的具体临床信息如表1所示。
药学论文参考
..................................
第四章总结与展望
本研究使用两种生物信息学分析方法(WGCNA分析和基于LASSO算法预后模型的构建分析)筛选出8个食管癌核心基因,并构建预测模型评价预后预测价值,结合临床预后数据最终确定ESCA的不良预后与KIF4A、CDKN3、CDT1、RRM2和MYBL2的过表达以及CELA2A、PGA3和PGA4的低表达有关。目前,食管癌基因靶向治疗仍然处于初始阶段,随着基因组学技术不断发展和进步,新的RNA被不断发现,使得食管癌相关分子机制被不断完善和发现,这对开发新的生物标志物和靶向治疗来提高食管癌患者的诊断率和生存率具有重大意义。同时本研究也存在一定的局限性。主要体现在食管癌预后基因的研究存在一定的异质性,这导致研究结果可能会存在相似的对医疗诊断参考的意义。这就意味着需要更大的临床样本来验证我们得到的数学模型。对于预后模型涉及具体癌基因或抑癌基因调控机制仍有待进一步研究。在本研究中,我们基于食管癌组织和癌旁组织差异表达基因的基础上构建模型,但TCGA数据库目前目前符合要求的样本较少,结合更多正常食管组织样本进行分析,是否可得到更优质模型仍待进一步挖掘。
参考文献(略)