本文是一篇医学论文,本研究发现与直肠腺癌的复发和转移密切相关主要为成纤维细胞生长因子的趋化、以微管和鞭毛为主的细胞骨架形成的调控相关生物过程、突触发生的调节以及细胞有丝分裂的调控密切相关生物过程、以及与细胞外基质的调控、微丝形成的调控的生物过程密切相关的生物过程;MAP4(cg04441191) 、 KSR2(cg05658717) 、 GRIN2A(cg09622330) 、YWHAG(cg10698404) 、 SPAG9(cg17047993) 、 CEP135(cg24504843) 、CEP250(cg24531267)七个基因的甲基化位点与直肠腺癌预后密切相关。
第1章 前言
1.1 结直肠癌的研究进展
肿瘤已成为威胁人类生命健康重要隐患,结直肠癌(CRC)作为目前世界上第三大最常见的癌症。 据统计,仅在 2017 年,美国就有超过 13 万名新的 CRC患者,其中有 50,000 多人因此而死亡[1]。在我国,CRC 的新发病例和致死病例近年来呈现逐年增加的趋势。2013 至 2018 年间,CRC 的发病率从 14.6/10 万人增长至 17.2/10 万人,致死率从 6.18/10 万人增长至 7.76/10 万人[2]。尽管,放化疗联合 TME 手术已成为局部晚期直肠癌(LARC)的标准治疗方法,并发现 LARC的局部控制率得到显著提高,但远处转移的风险和总体预后生存率并未得到明显改善,并且直肠癌的复发仍然是直肠癌患者死亡的主要原因[3]。因此,晚期 CRC治疗方案研究和 CRC 预后标志物的识别已成为癌症研究的热点问题。目前,针对 CRC 的研究主要集中于基于转录组水平上,采用差异表达基因分析算法(如SAM 算法、T 检验分析方法)结合 KM 生存分析,鉴定 CRC 的预后标志物或挖掘影响肿瘤增殖,迁移和侵袭密切相关基因和信号通路。
在转录组学水平上,Chao 等研究直肠腺癌样本(GSE35452)发现 DSG 在转录组水平上的表达与较差的肿瘤消退呈负相关,并与直肠腺癌的特异性生存、无局部复发生存,以及无转移生存呈显著负相关[4]。Millino 等研究发现 ABCC2基因和 miR-7,miR-182 等 6 个 miRNA 作为局部晚期直肠癌的术前放化疗响应靶标[5]。Nfonsam 等发现 MAPK 信号通路在发散性早发性直肠癌中出现显著失控,PI3K-AKT 信号通路在晚发性直肠癌中出现显著失控[5]。Horvat 等发现肿瘤位置与基因 APC 和 RASA1 突变显著相关、CRM 状态与 ATM 突变显著相关、淋巴结转移与 BRCA2 突变显著相关[6]。Wu 等发现 TIPE2 可以通过 Wnt /β-Catenin和 TGF-β/ Smad2 / 3 信号通路调节人直肠腺癌细胞的增殖,迁移和侵袭[5]。Song等对 GEO 数据集(GSE126095)分析,发现 SSTR2,CXCR5 和 SSTR3 的表达与患者的总体生存率密切相关[7]。Abdul Aziz NA 等应用 Illumina DASL 方法分析了 78 例 Duckes B 和 C 患者,发现了 19 个显着差异表达的基因,如 NOTCH2,ITPRIP,FRMD 等,这些基因与 CRC 的预后生存密切相关[1]。
....................................
1.2 DNA 甲基化研究进展
DNA 甲基化是指在 DNA 甲基转移酶的催化下,甲基(CH3-)从 S-腺苷甲硫氨酸供体转移到 DNA 中胞嘧啶碱基的 C5 位处的过程[8],其中,经过甲基修饰后的碱基叫做“5-甲基胞嘧啶”,简称为 5mC[9],经过修饰之后的 CpG 位点称为甲基化位点(Methylated loci)。DNA 甲基化代表着基本的表观遗传修饰,在细胞周期、细胞增殖、细胞凋亡、DNA 复制、染色质结构和基因转录等生物过程中扮演着重要作用。在许多疾病中,如癌症、退行性疾病以及衰老等都表现出异常的甲基化模式影响疾病表型的变化[10]。因此,以基因 DNA 甲基化水平的变化作为切入点探究疾病的分子分型或病理机制已成为表观遗传学中的研究热点。Wang 等应用 WGCNA 算法结合 lasso 回归算法发现 C20orf56, BTG2, C13orf16 等9 个基因的甲基化位点是肺腺癌的预后靶标[10]。Westerman 等应用 WGCNA 算法和 Comb-p 算法计算甲基化区域和甲基化模块,发现 SLC9A1,SLC1A5 和TNRC6C 甲基化与心血管疾病的患病风险密切相关[11]。
目前针对于 DNA 甲基化数据的研究主要集中于基于 DNA 甲基化位点识别方法的研究和基于 DNA 甲基化区域识别方法的研究。基于 DNA 甲基化位点识别方法的研究中,张等针对于 DNA 甲基化数据非正态分布和高异质性的特性,提 出 了 基 于 相 对 熵 的 差 异 甲 基 化 位 点 识 别 方 法 — — QDML (Quantitative identification of Differentially Methlated Loci),以解决数据因非正态分布而造成高假阳性率的问题,相比于 t-test、Wilcox、Combined t-test 三种传统差异分析方法,该方法具有高高检测能力、高精度,低假阳性率的特点[8],但该方法仍无法处理实验组和对照组数据未匹配的问题。
....................................
第2章 基于 DNA 甲基化的直肠腺癌病理机制研究及预后靶标识别
2.1 材料方法
数据分析流程如下:
医学论文怎么写
2.1.1 数据来源及数据预处理
TCGA 数据库(The Cancer Genome Atlas)(https://www.cancer.gov/)是由美国国家肿瘤研究所(National Human Genome Research Cancer Institute)和美国国家人类基因组研究所牵头联合 16个国家在 2006 年启动的目前最大的基因工程计划。研究者们希望通过大规模的基因组测序工作,构建人类全部癌症的基因组变异图谱,以挖掘癌症发生发展的主要机制,提供全新的癌症预防和治疗策略。截止至目前,TCGA 数据库已包含 40 大类肿瘤,32555 例癌症样本,3142246 种微小突变,以及所有样本的基因表达谱、CNV、DNA 甲基化、SNP、外显子测序结果、临床随访数据,以及肿瘤样本的免疫组化数据[22]。
本研究选择 TCGA 数据库中直肠腺癌的甲基化数据和临床样本数据作为研究对象,包括 103 个组织样本,其中,6 个癌旁组织样本和 97 个癌组织样本,接下来地,我们筛选甲基化样本与临床样本的交集样本,应用 stats R 包(v3.6.1)中的函数 hclust()进行层次聚类,去除离群样本,获得 84 个癌组织样本。最后,本研究去除在所有样本中β值小于 0的甲基化位点,获得 385475个甲基化位点。
................................
2.2 结果
2.2.1 差异甲基化基因的鉴定
本研究应用层次聚类算法去除离群样本,得到 90 个直肠腺癌样本的 DNA甲基化谱,其中包含 84 个癌组织样本、6 个癌旁组织样本(附图 1)。接下来地,我们去除零表达甲基化位点得到 385475 个甲基化位点。进而,我们应用 champ算法[34],以|log2FC|>0.235 & P < 0.05 为阈值,筛选得到 5983 个差异甲基化基因(19064 个差异甲基化位点)。
2.2.2 加权基因调控网络的构建和基因模块的鉴定
信号蛋白的遗传突变致使关键细胞信号传导的过度激活和因基因序列的表观遗传修饰导致抑癌基因转录的阻碍是肿瘤发生发展的重要途径[30]。Futreal 等研究表明在癌症中发生甲基化修饰和突变的基因多数参与细胞间的信号转导[31]。为此,本研究应用 Cui 等构建的信号调控网络作为基础[30],构建以差异甲基化基因信号调控网络作为背景网络,以所有基因 DNA 甲基化数据为背景数据,应用CCA 算法计算基因之间甲基化的相关性作为网络边的权重,得到一个包含 4719个基因的大网和若干个基因数小于 10 个小网,由此,我们定义大网为加权基因调控网络。接下来地,我们应用 mutilevel 算法对加权基因调控网络进行模块划分得到 20 个基因模块(modularity=0.59),去除基因数小于 100 的基因模块,获得16 个基因模块。
.............................
第 3 章 基于转录组学的直肠腺癌病理机制研究及预后靶标识别 .... 17
3.1 材料方法 .............................. 17
3.1.1 数据预处理和样本质控分析 ............................. 17
3.1.2 差异基因的识别 ........................... 18
第 4 章 基于拷贝数变异的直肠腺癌病理机制研究及预后靶标识别................30
4.1 材料方法 .............................. 30
4.1.1 原始数据预处理分析 ....................... 30
4.1.2 差异拷贝数变异基因鉴定 ............................ 31
第 5 章 影响直肠腺癌转移和复发的多组学特征基因分析 ................ 36
5.1 材料方法 .................................. 36
5.1.1 不同组学特征基因染色体分布情况分析 ...................... 36
5.2 结果 ............................... 36
第 5 章 影响直肠腺癌转移和复发的多组学特征基因分析
5.1 材料方法
5.1.1 不同组学特征基因染色体分布情况分析
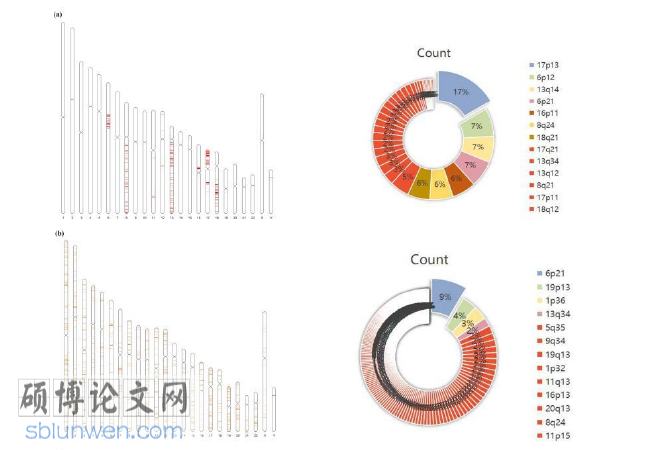
M、N、T 分期是评价实体瘤发生发展的重要指标[90]。为探究直肠腺癌转移和复发的关键机制,本研究首先对上述转录组水平、DNA 甲基化水平、拷贝数变异水平分析影响 M、N、T 表型变化的特征基因进行整合并分别取并集,得到不同组学影响直肠腺癌转移复发的特征基因群。进而探究不同组学特征基因在染色体上分布情况,本研究将不同组学特征基因群映射到染色体上,其中,基因信息注释应用R 函数getBM()实现,基因组信息可视化应用 R 函数ideogram()实现。
为探究不同组学中特征基因的分布情况,我们将在 DNA 甲基化水平上、拷贝数变异水平上、转录组水平上特征基因群比对至染色体上,发现在 DNA 甲基化水平上影响直肠腺癌的复发和转移的特征基因主要分布于 6p21 区域、19p13区域、1p36 区域、13q34 区域和 5q35 区域;在拷贝数变异水平上影响直肠腺癌的复发和转移的特征基因主要分布于 17p13 区域、6p12 区域、13q14 区域、6q21区域、16p11 区域、8q24 区域;在转录组水平上影响直肠腺癌复发和转移的特征基因主要分布于 3p21 区域、11q13 区域、17q25 区域、12q13 区域、19p13 区域(图 5.1)。
医学论文参考
...................................
第 6 章 结论
本研究针对直肠腺癌 DNA 甲基化芯片数据构建 DNA 甲基化加权基因调控网络模型,从 DNA 甲基化角度分析直肠腺癌的病理机制及预后靶标,接下来地,针对直肠腺癌转录组数据构建基于基因共表达网络结合社群发现算法及PageRank 算法的网络分析模型,从转录组角度探究影响直肠腺癌的复发和转移机制和预后靶标;为探究拷贝数变异水平上影响直肠腺癌转移和复发的关键机制,本研究针对直肠腺癌拷贝数变异数据构建基于随机森林的遗传算法探究直肠腺癌转移和复发机制;最后,本研究将 DNA 甲基化水平、转录组水平、拷贝数变异水平上特征基因映射到染色体上,探究影响直肠腺癌复发和转移的关键染色体区域。结论如下:
从 DNA 甲基化水平上分析,本研究发现与直肠腺癌的复发和转移密切相关主要为成纤维细胞生长因子的趋化、以微管和鞭毛为主的细胞骨架形成的调控相关生物过程、突触发生的调节以及细胞有丝分裂的调控密切相关生物过程、以及与细胞外基质的调控、微丝形成的调控的生物过程密切相关的生物过程;MAP4(cg04441191) 、 KSR2(cg05658717) 、 GRIN2A(cg09622330) 、YWHAG(cg10698404) 、 SPAG9(cg17047993) 、 CEP135(cg24504843) 、CEP250(cg24531267)七个基因的甲基化位点与直肠腺癌预后密切相关。
从转录组水平上分析,本研究发现细胞发育和分化,以及血管和神经系统的发育,两类生物学过程共同影响肿瘤的微环境,进而影响直肠腺癌的复发和转移。MMP14、SDC2、ACTA2、ZNF532,和 CYBRD1 在直肠腺癌的侵袭和转移上发挥重要作用,并与直肠腺癌的预后密切相关。
从拷贝数变异水平上分析,本研究发现影响临床表型 M 变化的特征基因主要参与线粒体相关的能量代谢、细胞对生长因子刺激的反应,以及细胞对肽激素刺激的应答反应;影响临床表型 N 变化的特征基因主要参与与突触相关的神经信号转导;影响临床表型 T 变化的特征基因主要参与与细胞间电信号的转导。MLIP、GCM1、GFRAL、GSTA3、MIR586 的拷贝数异常与直肠腺癌预后呈现非线性相关。
参考文献(略)