本文是一篇药学论文,本文设计合成了 1, 2, 4-噁二唑类化合物和 1, 3, 4-噁二唑类化合物。其中化合物 66显示出良好的 SHP2 抑制活性以及选择性,同时还有良好的细胞活性。
第一章 绪论
1.1 蛋白酪氨酸磷酸酶
蛋白酪氨酸磷酸酶(PTPs)是由人类中的 103 个基因编码,由包含 107 种信号酶组成的大家族,它们通过酶催化通过共价键形成中间体以达到催化底物的去磷酸化作用,涉及活性位点半胱氨酸残基的硫代磷酸酯键[1]。蛋白酪氨酸磷酸酶(PTPs)与蛋白酪氨酸激酶(PTKs)一起协同作用许多细胞活动,包括增殖、分化、存活、代谢和免疫反应等[2-3]。受到干扰的蛋白酪氨酸磷酸酶的活动会导致酪氨酸磷酸化异常,已被证明与多种人类疾病的病因有关,包括癌症,糖尿病和自身免疫功能障碍[4-6]。
目前,已有多种靶向 PTKs 的小分子抑制剂被 FDA 批准上市,而且还有许多小分子抑制剂正处于临床阶段。可惜的是尚未有靶向 PTPs 的抑制剂药物上市。虽然药物化学家们发现了一些靶向 PTPs 的抑制剂,但是由于 PTPs 催化域的高度同源性、生化机制复杂性、小分子的生物利用度低和细胞通透性差等问题。可喜的是许多 PTPs 家族酶可通过活性位点 Cys 的氧化、变构和低聚等各种调节机制调控人类疾病[4, 7]。
与 PTKs 相比,PTPs 的研究仍处于早期阶段,因为大多数 PTPs 家族酶的生化机制仍未研究清楚。随着对 PTPs 家族酶及其抑制剂的研究,以及信号通路机制的深入研究了解,进一步为相关疾病的治疗提供新的思路[8]。
..........................
1.2 SHP2 抑制剂研究进展
SHP2 是由 PTPN11 编码的包含 Src 同源 2 域的非受体型蛋白酪氨酸磷酸酶。SHP2是包括 593 个氨基酸残基,有两个 SH2 域(N-SH2 和 C-SH2),一个 PTP 催化域,一个具有四个酪氨酸磷酸化位点(Tyr542,Tyr546,Tyr580 和 Tyr584)和一个脯氨酸富集区。晶体结构显示,两个 SH2 域的中心均包含多个 β 折叠,每侧均包含一个 α 螺旋。在 PTP域中具有一些 α 螺旋和 β 折叠的混合体结构。两个 SH2 域都围绕着 PTP 域。N-SH2 域中的 βD'-D'E-βE 与 PTP 域催化口袋中的磷酸酪氨酰(pTyr)环、P 环和 Q 环相互作用,从而抑制了催化部位。C-SH2 结构域的每个末端都连接到上述两个结构域,没有明显的相互作用,但其相对方向使整体结构呈现封闭结构。在 SH2 结构域的远离 PTP 结构域的表面上有完全暴露的 pTyr 肽的结合位点(图 1-1)[9-10]。在正常情况下,SHP2 通过 N-SH2 结构域和 PTP 催化口袋形成分子内氢键使其处于自抑制状态[10-11]。但是,在受到刺激后,N-SH2 结构域远离 PTP 结构域。然后将 SHP2 的构象更改为开放状态,并将 PTP催化口袋暴露,发挥生物学功能。
位于细胞质中的 SHP2 可以被各种受体酪氨酸激酶募集来诱导细胞信号传导,并参与多个细胞内致癌信号传导级联反应,例如 Jak / STAT[12-13]、PI3K / AKT[14-15]、RAS / Raf / MAPK[16-18]、PD-1 / PD-L1[19-20]和 mTOR 通路[21]。然而,在几种人类疾病中发现 SHP2的失控(包括已识别的突变,过度表达和下调)会导致底物以及结合伴侣的催化活性和结合亲和力发生变化[22]。SHP2 的这些不同状态可能与复杂的病理过程密切相关。
SHP2 在胚胎和成年动物中普遍表达,在调节细胞生长、分化、存活和凋亡中起着重要的信号转导作用[23-24]。研究表明,SHP2 是正常发育和造血所必需的[25-26]。它的失调会导致各种人类疾病,包括 Noonan 综合征(NS),Leopard 综合征(LS),白血病和实体瘤等[22, 27-28]。在疾病 Noonan 综合征和 Leopard 综合征中 PTPN11 会发生高频率突变,同时在白血病和实体瘤中的 SHP2 被发现作为肿瘤启动子。目前 SHP2 是一种潜在的癌症治疗靶点。在过去的二十年中,小分子 SHP2 抑制剂引起了广泛的关注。然而,PTPs 家族催化域的高度同源性使 SHP2 抑制剂的开发存在巨大挑战,同时还需要提高小分子抑制剂的细胞渗透性和生物利用度[11]。因此,迫切需要一些新颖有效选择性高的SHP2 小分子抑制剂。
..........................
第二章 材料与方法
2.1 实验材料
2.1.1 实验试剂
苯甲腈,呋喃-2-腈,噻吩-2-腈,吡啶-3-腈,1H-吲哚-2-腈,4-硝基苯腈,3-硝基苯腈,3,4-二氯苯腈,2,3-二氯苯腈,4-氯-3-氟苯腈,3-氯-2-氟苯腈,4-溴-3-硝基苯腈,4-乙炔基-3-硝基苯腈,2-氯-3-硝基苯腈,3-氯-4-硝基苯腈,4-硝基-3-三氟甲基苯腈,3-甲氨基-4-硝基苯腈,苯甲酸,2-呋喃甲酸,2-噻吩甲酸,4-甲苯甲酸,3-甲苯甲酸,3-氯苯甲酸,3-硝基苯甲酸,4-甲氨基苯甲酸,4-甲氧基苯甲酸,3-甲氧基苯甲酸,3,4-二甲氧基苯甲酸,4-氟-3-硝基苯甲酸,4-甲基-3-硝基苯甲酸,4-乙基-3-硝基苯甲酸,4-甲氧基-3-硝基苯甲酸,盐酸羟胺,甲胺,异丙胺,锌粉,2-(7-氮杂苯并三氮唑)-N,N,N',N'-四甲基脲六氟磷酸酯(HATU),1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC),氮气,氯化钠,碳酸氢钠,碳酸钾,氢氧化钠,氢氧化钾,无水硫酸镁,氯化亚锡二水合物,乙醇钠,N,N-二异丙基乙胺(DIPEA),盐酸,二氯亚砜,三氯氧磷,二甲基亚砜,草酸二乙酯,水合肼,N,N-二甲基甲酰胺(DMF),N,N-二甲基乙酰胺(DMA),乙酸,二氯甲烷,乙酸乙酯,石油醚,甲醇,乙醇。(实验溶剂和试剂都是通过商业途径购买得到,主要由上海化学试剂公司提供。)
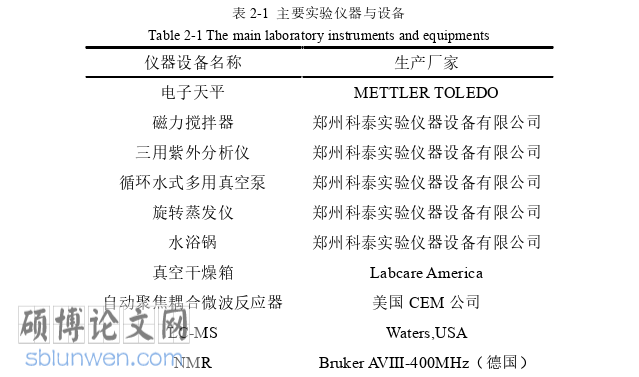
2.1.2 实验仪器与设备
化学合成、化合物结构分析与检测所需要的主要仪器和设备
药学论文怎么写
............................
2.2 实验方法
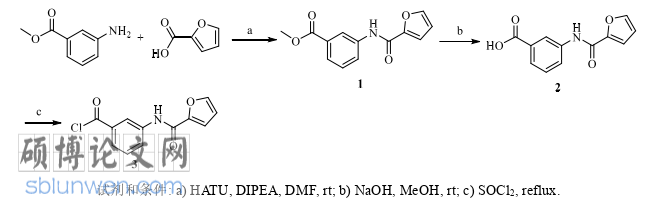
反应操作 1
药学论文参考
将 2-呋喃甲酸(1.0 eq)溶解在 DMF 中,向其加入 HATU(1.2 eq)和 DIPEA(2.0 eq),氮气保护下反应 1 小时后向反应中加入间氨基苯甲酸甲酯(1.1 eq),氮气保护下过夜。反应完成后加入大量乙酸乙酯,使用 1 M HCl 洗涤两次,再使用饱和碳酸氢钠洗涤一次,最后使用饱和氯化钠洗涤至有机相无 DMF 残留,干燥有机相并减压浓缩得化合物 1。将化合物 1(1.0 eq)溶解在甲醇中,向其加入氢氧化钠(5 eq)常温下过夜。反应完成后减压浓缩,加入蒸馏水溶解,使用 1M HCl 使溶液至中性,抽滤并干燥滤渣得得化合物 2。将化合物 2 加入到 SOCl2 中,N2 保护下回流三小时。反应完成后减压浓缩得化合物 3。
.........................
第三章 结果与讨论 ........................... 21
3.1 含 1, 2, 4-噁二唑结构单元抑制剂的设计合成及生物活性评价 ........................... 21
3.1.1 含 1, 2, 4-噁二唑结构单元抑制剂的设计 .................................. 21
3.1.2 含 1, 2, 4-噁二唑结构单元抑制剂的合成与表征 ........................................ 21
主要结论与展望.........................30
主要结论........................30
展望........................30
第三章 结果与讨论
3.1 含 1, 2, 4-噁二唑结构单元抑制剂的设计合成及生物活性评价
3.1.1 含 1, 2, 4-噁二唑结构单元抑制剂的设计
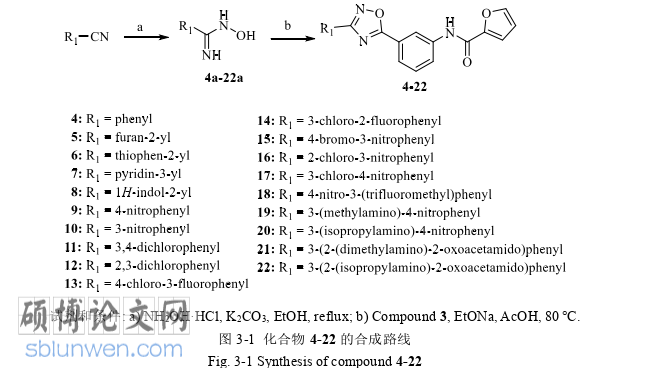
在含 1, 2, 4-噁二唑结构单元抑制剂的设计之前,我们对本课题组之前合成的化合物进行总结并分析构效关系,通过骨架跃迁设计出一系列含 1, 2, 4-噁二唑结构单元抑制剂。首先通过逆合成分析我们首先合成了重要中间体化合物 3,随后使用氰基化合物在盐酸羟胺作用下得到酰胺肟,随后与化合物 3 合环得到化合物 4-22。随后我们根据活性测试结果,总结构效关系,增加化合物多样性,设计合成出了化合物 26-41。最后,进一步衍生化,设计合成出了化合物 45-48。
3.1.2 含 1, 2, 4-噁二唑结构单元抑制剂的合成与表征
3.1.2.1 含 1, 2, 4-噁二唑结构单元化合物 4-22 的合成与表征
药学论文参考
...........................
主要结论与展望
主要结论
本课题组筛选出以含 2,3-二氢萘嵌间二氮杂苯结构单元的阳性化合物为研究基础,设计并合成了靶向 PTPs 家族多种化合物库。我们通过其中 SHP2 抑制剂杂环双芳基酰胺化合物库,进行骨架跃迁得到噁二唑化合物。
首先以 1, 2, 4-噁二唑为母核,首先对结构中 1, 2, 4-噁二唑的 3 位引入各种杂环和含有不同取代基的苯环,设计合成得到 19 个 1, 2, 4-噁二唑类化合物,并对其进行 SHP2 蛋白抑制实验。结果表明含有硝基的化合物 10(IC50 = 10.44±0.37 μM)对 SHP2 蛋白显示出一定的抑制活性。此外,进一步设计合成了一系列硝基取代化合物,结果发现化合物19(IC50 = 7.94±0.71 μM)表现出较好的 SHP2 抑制活性。基于此进一步分子结构衍生化,得到化合物 35(IC50 = 42.12±3.11 μM)和化合物 37(IC50 = 9.94±0.17 μM)。
总结 1, 2, 4-噁二唑的抑制实验结果,本论文设计合成了一系列 1, 3, 4-噁二唑类化合物。首先固定 3-(呋喃-2-甲酰胺基)-苯基基团筛选得到有较好活性的化合物 63(IC50 = 8.00±0.85 μM)。进一步衍生化得到化合物 66(IC50 = 2.73±0.20 μM)、化合物 68(IC50 = 11.35±0.68 μM)、化合物 70(IC50 = 12.92±1.23 μM)和化合物 77(IC50 = 24.60±0.40 μM)有比较好的 SHP2 抑制活性,其中化合物 66 的活性较为优异。1, 3, 4-噁二唑类化合物的SHP2 蛋白抑制实验结果表明,4-甲氨基-3-硝基苯基基团和 3(-呋喃-2-甲酰胺基)-苯基基团在该类活性抑制剂中会起到十分重要的作用。进一步对化合物 66 在 TF-1 细胞中进行了抗增殖实验,结果发现化合物 66 抑制了 SHP2 蛋白从而影响了下游信号通路,抑制了 TF-1 细胞增殖(IC50 = 6.67±0.58 μM)。这可以对于后续 SHP2 蛋白抑制剂的设计提供了新思路。最后进行了分子动力学模拟,发现化合物 66 的 4-甲氨基-3-硝基苯基插入到了 H172 和 H171 所形成的口袋,可以增强化合物 66 与 SHP2 蛋白的相互作用。分子动力学模拟结果在某种程度上可以解释化合物 66 对 SHP2 的良好抑制活性的原因。
参考文献(略)